Large granular lymphocytes (LGL) comprise 10 – 15 % of lymphocytes in blood in healthy adults (1). Most of these lymphocytes (85 %) are CD3– natural killer (NK) cells and the rest are CD3+ cytotoxic T cells. LGLs are medium-sized round or oval cells with eccentrically placed nuclei with condensed nuclear chromatin without a clear nucleolus and with surplus cytoplasma (pale blue) containing a few large azurophilic granules (fig 1).
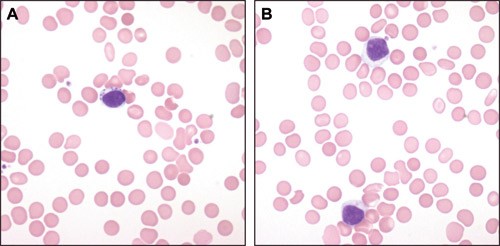
Figure 1 Two blood smear specimens from a patient with LGL-leukaemia. a) A large lymphocyte with pale blue cytoplasm and 7 – 8 large and powerful azurophilic granules. b) Two large lymphocytes with a surplus of pale blue cytoplasm with very discrete azurophilic granules
LGL-leukaemia was first described in 1985, as a clonal disease with affection of blood, spleen and bone marrow (2). Several years earlier, Hovig and collaborators had described a patient with rheumatoid arthritis, splenomegaly, neutropenia and LGL. In 1975, Brouet and collaborators found that in six of 11 patients with chronic lymphatic leukaemia (T-cell type) the lymphocytes had large amounts of cytoplasm with azurophilic granules (3, 4). The link to rheumatoid arthritis and neutropenia became clear a couple of years later (5).
In 1993, Loughran described two types of LGL-leukaemia: one T-cell type which comprised the majority of cases and a much rarer NK-cell type (6). This dichotomy is adapted by the WHO classification. T-LGL-leukaemia is almost always a very indolent disease, while NK-LGL-leukaemia often has an aggressive clinical phenotype. Aggressive variants of T-LGL-leukaemia have been described. Indolent NK-LGL-leukaemia also occurs, but it is difficult to show that NK cells are clonal. It is therefore very challenging to decide with certainty whether persisting lymphocytosis of NK cells really is an expression for NK-LGL-leukaemia. In such cases, clinical symptoms and infiltration of NK cells in liver, spleen, bone marrow and possibly other organs are much emphasised.
LGL-leukaemia has been perceived as a very rare disease (7, 8). LGL-leukaemia comprises 2 – 5 % of all T-NK-lymphoproliferative diseases and up to the year 2003, only 400 cases have been described in the literature (8). During the last 6 years, our experience indicates that LGL-leukaemia is not as rare as it may seem from the literature, and that the disease is underdiagnosed. The aim with this study is to direct attention to the disease and show its clinical phenotypes, discuss diagnostic possibilities and challenges and present treatment indications and treatment alternatives. Patients with LGL-leukaemia may be encountered in the primary health services and in the specialist services. Knowledge of the disease may contribute to a targeted and quick diagnosis and ensure that effective treatment may be offered to patients in need of treatment.
Material and methods
Patients
The study included all patients for whom Rikshospitalet (Section for blood diseases, Medical department) had been involved in diagnosing them with LGL-leukaemia in the period 1.10.2001 – 31.12.2007. The diagnostic criteria we used were (8):
Persisting expansion of LGL in blood and/or infiltration of LGL in bone marrow or extramedullary tissue
Characteristic immunophenotype assessed by flow cytometry or immunocytology (CD3+CD16+CD56+/–CD57+ or CD–3CD16/56+CD57+)
Clonal T/NK-cell populations with clonally rearranged T-cell receptor (TCR) as determined by molecular genetic methods, or TCR-Vβ-restriction as determined by flow cytometry and/or detection of clonal cytogenetic deviation by karyotyping
Clinical symptoms, seen in 70 – 75 % of the patients, are also emphasised in the diagnostic process and clinical symptoms are of special importance in the assessment of whether there is an indication for treatment or not
All the first three criteria must be fulfilled to make the diagnosis LGL-leukaemia.
Method
Clinical information from all patients who had been diagnosed with LGL-leukaemia (for whom Rikshospitalet [Section for blood diseases, Medical department] had been involved in the diagnosis), was retrieved from medical records and laboratory data at the hospital (by using the patient administrative system and ICD-10 codes) and reviewed.
We do not have systematic follow-up data for all patients, as those who were not examined clinically at Rikshospitalet were not always examined systematically with respect to clinical chemistry and immunological analyses that are useful to shed light on phenotypical traits, but not strictly necessary to diagnose the disease.
Results
Incidence
In the 6-year study period, we have diagnosed LGL-leukaemia in 52 patients; 26 women and 26 men (tab 1). The median age at diagnosis was 59 years (26 – 86 years). The patients were not exclusively from the South-Eastern Regional Health Authority, and we cannot be certain that we have included all patients from the area of recruitment who have been diagnosed with LGL-leukaemia in the study period. Therefore, the material does not enable us to calculate an incidence for LGL-leukaemia for all of Norway.
|
Table 1 Characteristics of 52 patients with LGL-leukaemia
|
|
Number
|
52
|
|
Women
|
26
|
|
Men
|
26
|
|
Haematology
|
|
|
Neutrophilic granulocytes (n = 41)
|
|
Median (spread) · 10⁹/l
|
1.5 (0 – 11.4)
|
|
< 1.0 · 10⁹/l
|
18
|
|
< 0.5 · 10⁹/l
|
15
|
|
Thrombocytes (n = 43)
|
|
|
Median (spread) x 10⁹/l
|
198 (42 – 456)
|
|
< 150 · 10⁹/l
|
8
|
|
< 100 · 10⁹/l
|
3
|
|
Haemoglobin (n = 42)
|
|
|
Median (spread) g/100 ml
|
12.8 (6.4 – 16.8)
|
|
< 11 g/100 ml
|
9
|
|
< 10 g/100 ml
|
4
|
|
Lymphocytes (n = 37)
|
|
|
Median (spread) x 10⁹/l
|
2.6 (0.6 – 247)
|
|
Organomegaly
|
|
|
Splenomegaly
|
10
|
|
Hepatomegaly
|
4
|
|
Splenectomy
|
8
|
|
Other disease
|
|
|
Rheumatoid arthritis
|
11
|
|
Systemic lupus erythematosus
|
1
|
|
Myositis
|
1
|
|
Polyneuropathy
|
2
|
|
Other clonal lymphoproliferative disease
|
|
Monoclonal gammopathy
|
8
|
|
Lymphoma
|
1
|
|
Other malignant disease
|
|
|
Cancer
|
7
|
|
Myelodysplastic syndrome
|
1
|
Immunophenotype and clonality
All patients, with one exception, had LGL-leukaemia of the T-cell type. In 48 patients the leukaemia cells had immunophenotypes characteristic of cytotoxic T cells (CD2+CD7+CD3+CD8+CD16+CD57+) (fig 2), but in one patient (who had an aggressive clinical phenotype) the immunophenotype was CD2+CD7+CD3+CD8+CD57–; for two patients who had the immunophenotype given above the leukaemia cells were CD4+CD8–. A clonally rearranged T-cell receptor was documented in all patients. In one patient the leukaemic NK cells had the immunophenotype; CD2+CD7+CD3–CD8+ CD57+. This patient had persisting lymphocytosis (> 5 x 10⁹/l) over several years, and we have documented infiltration of lymphocytes with the particular immunophenotype in biopsies from bone marrow, gastrointestinal tract and skin. We believe that the diagnosis NK-LGL-leukaemia is well founded in this patient, despite the fact that we have not identified the cells as clonal (cytogenetic examination).
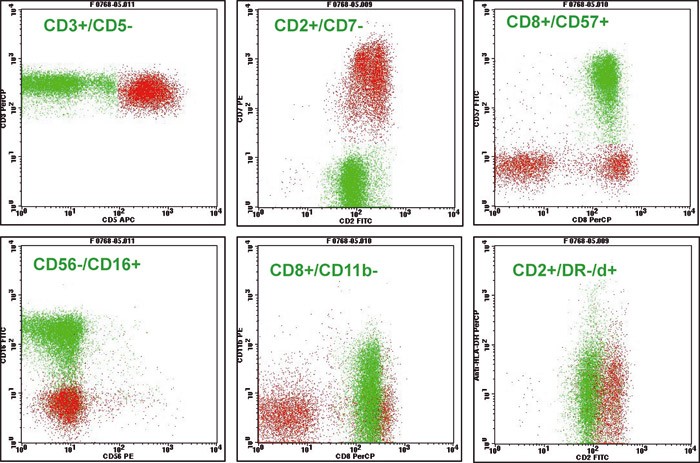
Figure 2 A flow cytometric examination of lymphocytes in blood in patients with LGL- leukaemia. The clonal T-cell population is found as the green population in the cytograms and has the following immune phenotype: CD2+CD3+CD8+CD16+CD57+. Non- expression of CD5 and CD7 on the leukaemic T cells are common with LGL-leukaemia of the T-cell type
Bone marrow morphology
of the bone marrow cells (fig 3), and intrasinusoidal localisation was common. Lymphoid infiltrates were also common, and they consisted of polyclonal B-cells surrounded by a rim of polyclonal CD4+ T-cells.
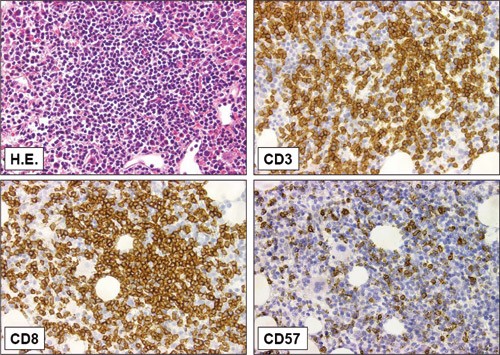
Figure 3 Trephine biopsy from a patient with LGL-leukaemia with extensive bone marrow affection. A specimen stained with haematoxylin and eosin (H.E.) shows a diffuse infiltration of small and medium-sized lymphoid cells. Most of the lymphocytes are CD3+ and CD8+, and many express the NK-cell antigen CD57 (x40)
All patients had a trilinear haematopoesis with a normal thrombocytopoesis. The erythropoesis was normal in most patients, but it was increased in patients with haemolytic anaemia and strongly reduced in patients with erythroaplasia. The granulocytopoesis was usually normal, but it was reduced with signs of maturation block in some patients with granluocytopenia.
Clinical presentation
Patients requested help from the health services for many reasons. Most of them had symptoms and clinical findings such as repeated infections, autoimmune disease and splenomegaly, or laboratory findings in the form of cytopenias (tab 1, tab 2). The most common finding was granulocytopenia, usually but not always concomitant with infections (26 patients totally [50 %]), 18 [35 %] with infections). Anaemia was the main problem in five patients (10 %), and the cause of the anaemia was either haemolysis (negative finding in direct antiglobulin test [DAT]) or erythroplasia. One patient had the diagnosis T-LGL-leukaemia after an assessment for thrombocytopenia, but thrombocytopenia was found in totally 11 patients. No patient had serious thrombocytopenia with bleeding manifestations.
|
Table 2 Haematological parameters (median, spread) in patients with LGL-leukaemia by clinical presentation
|
|
Neutropenia (n = 26)
|
Anaemia (n = 5)
|
Thrombocytopenia (n = 1)
|
Lymphocytosis (n = 9)
|
|
Haemoglobin (g/100 ml)
|
12.2 (7.3 – 17)
|
8.9 (6.4 – 11.3)
|
12.3
|
14.3 (12.1 – 16.8)
|
|
Leukocytes (· 10⁹/l)
|
2.7 (0.8 – 18.6)
|
8.95 (2.7 – 13.5)
|
4.06
|
12.3 (8.7 – 453)
|
|
Granulocytes (· 10⁹/l)
|
0.2 (0 – 3.8)
|
1.5 (0.86 – 2.0)
|
2.07
|
3.2 (< 1 – 11.41)
|
|
Lymphocytes (· 10⁹/l)
|
1.46 (0.64 – 13.4)
|
6.1 (1.42 – 10.78)
|
1.47
|
8.8 (4.9 – 247)
|
|
Thrombocytes (· 10⁹/l)
|
191 (99 – 414)
|
315 (158 – 456)
|
68
|
198 (42 – 333)
|
In eight patients (15 %), the assessment of moderate lymphocytosis led to the diagnosis, while for one patient the diagnosis was based on the presence of general symptoms, splenomegaly, discrete lymphadenopathy and excessive lymphocytosis. 19 patients (37 %) were asymptomatic at the time of the diagnosis, at least regarding symptoms that can reasonably be related to LGL-leukaemia, and their cytopenias or other indications of the diagnosis (for example lymphocytosis) were uncovered bycoincidence. Ten patients (19 %) had splenomegaly. In addition, eight patients had splenectomies performed before being diagnosed with LGL-leukaemia. We have sparse information about the indication for the splenectomy in these patients, but there are reasons to believe that several of them had splenomegaly. Four patients (8) had hepatomegaly, and two patients (4 %) had a discrete, but general lymphadenopathy.
Many patients had other diseases with immune pathogenesis (tab 1). Rheumathoid arthritis was the most frequent cause of comorbidity (21 %). Ten of the 29 examined patients tested positive for rheumatoid factor, and three of the 19 examined patients had positive findings in the direct antiglobulin test.
Signs of other clonal lymphoprolipherative disease, usually monoclonal gammopathy, were found in nine patients (tab 1). In patients with monoclonal gammopathy we could not give a definite diagnosis of malignancy, and these cases must be categorised as MGUS (monoclonal gammopathies of undetermined significance). The result of the protein electrophoresis of serum was only known in 18 patients. Eight patients (15 %) had non-lymphoid malignancies, seven were diagnosed with cancer and one with myelodysplasia. These diagnoses were made before or during follow-up of the LGL-leukaemia.
Treatment
The indication for starting treatment was cytopenia with associated symptoms (tab 3) for all patients except one who had NK-LGL-leukaemia and for whom general symptoms and skin affection were the main reason for treatment. The patients were treated with cyclosporine or methotrexate. For the cyclosporine treatment the goal was to achieve a cyclosporine trough concentration of 100 – 150 µg/l. Upon effect, the cyclosporine dose was reduced to the lowest possible effective dose to reduce the risk of nephrotoxicity upon long-term treatment. Osuji and collaborators have described the response criteria for this type of treatment (9). Methotrexate was given orally once weekly (10 mg/m²), and the dose was adjusted according to effect and/or possible adverse events. Cyclosporine was combined with G-CSF (granulocyte colony-stimulating factor) in a few patients who did not have the intended effect of cyclosporine monotherapy (tab 3). The combination treatment turned out to be effective, and we could discontinue G-CSF and continue with cyclosporine alone with acceptable effect.
|
Table 2 Haematological parameters (median, spread) in patients with LGL-leukaemia by clinical presentation
|
|
Neutropenia (n = 26)
|
Anaemia (n = 5)
|
Thrombocytopenia (n = 1)
|
Lymphocytosis (n = 9)
|
|
Haemoglobin (g/100 ml)
|
12.2 (7.3 – 17)
|
8.9 (6.4 – 11.3)
|
12.3
|
14.3 (12.1 – 16.8)
|
|
Leukocytes (· 10⁹/l)
|
2.7 (0.8 – 18.6)
|
8.95 (2.7 – 13.5)
|
4.06
|
12.3 (8.7 – 453)
|
|
Granulocytes (· 10⁹/l)
|
0.2 (0 – 3.8)
|
1.5 (0.86 – 2.0)
|
2.07
|
3.2 (< 1 – 11.41)
|
|
Lymphocytes (· 10⁹/l)
|
1.46 (0.64 – 13.4)
|
6.1 (1.42 – 10.78)
|
1.47
|
8.8 (4.9 – 247)
|
|
Thrombocytes (· 10⁹/l)
|
191 (99 – 414)
|
315 (158 – 456)
|
68
|
198 (42 – 333)
|
In two patients with erythroplasia, erythropoietin was used in addition to cyclosporine. One of the patients did not need transfusions after the combination treatment, while the other patient did not respond and was successfully treated with pentostatin (4 mg/m² intravenously every 2 week). Cyclosporine was discontinued because of nephrotoxicity in two patients and replaced with methotrexate with a persisting effect. One patient received G-CSF as monotherapy, with a good effect. The reason was that the patient’s granulocytopenia had been regarded as chronic idiopatic neutropenia for a long time.
The patient who had an aggressive disease with clear clinical similarities with T-PLL (T-prolymphocyte leukemia), was initially treated with alemtuzumab followed by allogen stem cell transplantation with an HLA-identical family donor during first remission.
Discussion
We have diagnosed LGL-leukaemia in 52 patients over a 6-year period. If LGL- leukaemia only comprises 2 – 5 % of all T/NK-lymphoproliferative diseases, as indicated by the literature (8), this is a surprisingly high number. We believe that the disease is probably underdiagnosed. The prognosis is good (median survival 14.5 years) if the cytopenia is treated (also in patients who have serious granulocytopenia [9]), and survival is probably similar to that in an age-adjusted control group. This implies that the disease prevalence has a certain magnitude.
In our material there were similar numbers of men and women and the median age at diagnosis was 59 years. The clinical symptoms in our patients were almost exclusively related to cytopenias, and neutropenia with repeated infections was the most common clinical phenotype. In our patients, we also found a high prevalence of other diseases with immunopathogenesis. Our material corresponds well with the literature in this respect (6 – 9).
MGUS was identified in eight patients, i.e. 44 % of patients for whom the result of protein electrophoresis was known to us and 15 % of the entire material. The prevalence of MGUS in an unselected Caucasian population with the same median age is about 3 % (10). Viny and collaborators have recently reported a corresponding high prevalence of clonal B-cell disease in LGL-leukaemia (11).
Cytopenias in adult patients without a clear cause, should raise suspicion of LGL-leukaemia (11). Some have questioned the legitimacy of this diagnosis; in our opinion it is useful even if we agree that the term leukaemia may be challenging when informing patients. Firstly, the diagnosis can be the end of long-winded diagnostic efforts. Secondly, the diagnosis may lead to effective treatment when indicated.
Drug treatment, in the form of cyclosporine or methotrexate, is not cytoreductive but immunomodulating. However, one may see a reduction of the number of tumour cells in blood and bone marrow with methotrexate treatment, but this is not regarded as essential for the treatment effect. We do not have exhaustive information about treatment results in this patient material, but treatment results for eight of the patients have been published earlier as part of a larger international multicentre study (9). Cyclosporine and methotrexate are regarded as equal treatment alternatives, and response is expected in 80 % of the patients (9). In a small minority of patients it may be indicated to combine the immunomodulating treatment with G-CSF or erythropoietin. The effect is usually sustained with persistent immunomodulating treatment, even if the cytokine treatment is discontinued (9). The immunomodulating treatment can usually not be discontinued without loss of effect even if the remission may persist for years after end of treatment in a few patients (9). Powerful cytoreductive treatment will often prove to be ineffective (12). No published results are available that recommend treatment in those rare patients where LGL-leukaemia have an aggressive phenotype. The presentation in one of our patients had much in common with T-PLL, and we therefore chose a treatment strategy as for this condition (13).
At least a third of patients were asymptomatic, and especially in these one may naturally ask whether it was right to diagnose them with LGL-leukaemia. It is possible that the term MTL (monoclonal T-lymphocytosis), which so far has not been launched internationally, would be at least as suitable, and at least as easy to relate to for the patients. MBL (monoclonal B-lymphocytosis) has been launched as a term in cases where there is a clonal expansion of mature B-lymphocytes, but only in cases where the diagnostic demands for chronic lymphatic leukaemia or other chronic lymphoprolipherative disease have not been met (14). LGL-leukaemia of the indolent type seldom or never shows any substantial progression as a tumour disease, and transformation to an aggressive prolipherative disease is rare (15). Therefore, there are arguments for using the MTL term instead of T-LGL- leukaemia, especially in cases where patients lack clinical manifestations.
