Left-ventricular noncompaction (LVNC) has been reported in fewer than 1 % of patients in various echocardiographic studies, and may exist as a familial disposition. The condition arises from an arrest in normal myocardial embryogenesis (1) and has recently been described as a cause of left-ventricular dysfunction (1 – 6). Affected children and adults may develop heart failure, thromboembolic events, and/or ventricular arrhythmias (1, 2, 7 – 9). Two-dimensional echocardiography or magnetic resonance imaging (MRI) of the heart is used to diagnose the condition (5, 7, 9 – 12). Typical findings include a thin, normally developed outer layer of the left- ventricular myocardium, and an inner layer (closest to the endocardium) thickened by trabeculae with deep intertrabecular recesses (fig 1).
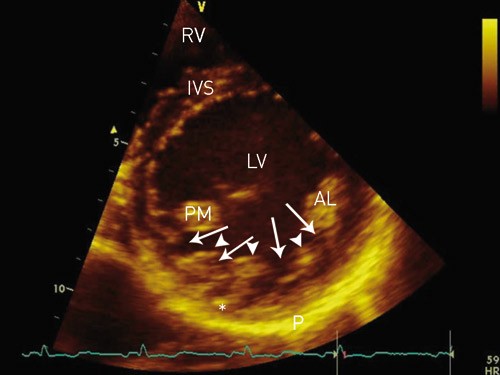
Figure 1. Transthoracic echocardiography, parasternal short axis view of the LV. The inferolateral wall of the LV with prominent trabeculae (arrowheads) and deep intertrabecular recesses (arrows). The normally developed epicardial myocardial layer (star) in the region is notably thinner than normal, when compared with the thickness of IVS. AL = anterolateral papillary muscle, IVS = interventricular septum, LV = left ventricle, RV = right ventricle, P = pericardium, PM = posteromedial papillary muscle
Material and methods
Articles from our own literature archive and relevant references in them were reviewed.
Morphology and diagnosis
During embryogenesis, the mesh of muscle fibres making up the heart normally becomes compacted while the intertrabecular sinusoids degenerate. This compaction process occurs in gestational weeks 5 – 8, and results in transformation of intertrabecular sinusoids to capillaries. It starts in the epicardium and progresses inward towards the endocardium, from the base of the heart to apex, and is more complete in the left ventricle (LV) than in the right ventricle (RV). Development of the coronary circulation occurs in the same period (1, 13) and is inhibited in the presence of noncompaction. The result is multiple persistent and prominent ventricular trabeculae, and deep intertrabecular recesses which communicate with the ventricular cavity and not with the coronary circulation (fig 1, fig 2). The ventricular cavity and the intertrabecular recesses are covered by a continuous endothelium (1, 2, 5). Predilection sites for noncompaction (normal findings in fish, amphibian and reptile hearts [4]) are the mid and distal segments of the lateral and inferior walls, as well as in the apical region (5, 9, 13). The extent of changes varies among patients (1, 4).
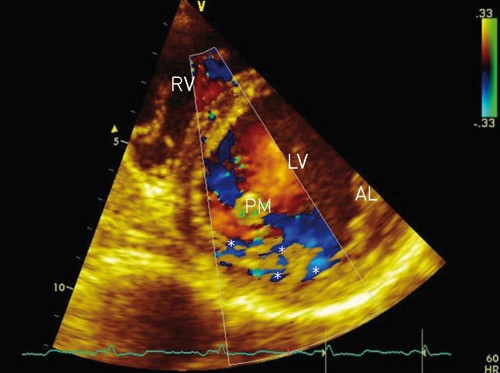
Figure 2. Transthoracic echocardiography, parasternal short axis view of the LV. Colour Doppler of blood flow in the LV shows blood flow (star) in the interventricular recesses. AL = anterolateral papillary muscle, LV = left ventricle, PM = posteromedial papillary muscle, RV = right ventricle
Literature presents the condition with many names; isolated LVNC, honeycombed myocardium, spongy myocardium, persisting myocardial sinusoids, myocardial disorganization and left- ventricular hypertrabeculation (4, 7). In principle they all describe the same condition.
LVNC was first described in 1932, in connection with autopsy of a newborn child who had aortic atresia and a coronary-ventricular fistula (14). Several publications and review articles have described the condition since then (1–6, 15, 16). LVNC may present concomitantly with other congenital anomalies of the heart (such as ventricular septal defects or ventricular outflow obstruction), and isolated (without other congenital anomalies of the heart) with or without other rare extracardiac conditions (such as systemic myopathies or facial dysmorphism) (2–7, 9–11). The isolated type without extracardiac abnormalities will be discussed in the following, unless otherwise specified.
Increased knowledge about the pathogenesis, diagnostic characteristics and prognosis of LVNC has recently caused the condition to be classified as primary cardiomyopathy (17). Echocardiography or MRI of the heart (12) are considered to be diagnostic gold standards.
Several echocardiographic findings are considered to be typical (2, 5, 7, 9 – 11):
Absence of other cardiac anomalies (by definition).
Involvement of the apex and/or apical and/or midventricular segments of the inferior and/or lateral wall. More than 80 % of patients with LVNC display noncompaction in one or more of these predilection sites.
Three or more prominent myocardial trabeculae in the same image plane.
Deep intertrabecular recesses that communicate with the ventricular cavity.
The affected myocardial segments have two layers: a thin epicardial layer and a thickened trabecular endocardial layer.
The outer myocardial layer is compacted in a normal way, but the inner layer is noncompacted. When the wall segments are measured at end-systole, the relationship between the noncompacted layer and the compacted layer will typically be > 2.0.
There is no international consensus about strict diagnostic criteria for LVNC and one or more of the echocardiographic criteria listed above may also be found in other conditions or diseases that affect the LV. However, presence of all the above-mentioned criteria is reported to be highly specific for LVNC (11). When findings are equivocal, intravenous echocardiographic contrast agents may help to make the diagnosis (18). In magnetic resonance images of the heart, a noncompacted/compacted layer ratio ≥ 2.3 (measured in the ventricle’s end diastole) is considered typical for LVNC (12) (fig 3). Other cardiac imaging modalities such as computer tomography and ventriculography may also be useful (19).
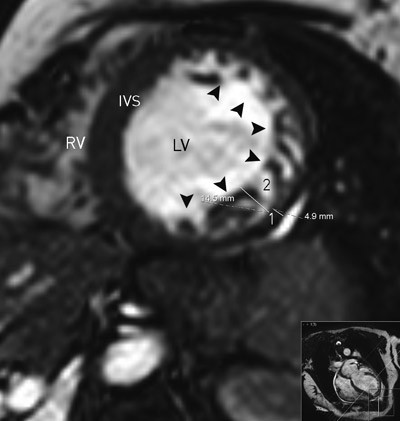
Figure 3. Magnetic resonance tomography of the heart. End-diastolic short axis view of the LV and RV. The lateral wall and adjacent segments of the inferior and anterior wall (arrowheads) with noncompaction changes, including a thinner epicardial layer (measurement 1: 4.9 mm) and a thicker trabecularized endocardial layer (measurement 2: 14.5 mm). End-diastolic thickness of the inner layer is 3.0 times that of the outer layer. LV = left ventricle, RV = right ventricle
Obvious trabeculation is a normal finding in the RV. However, in LVNC up to half of cases also have more pronounced trabeculation of the RV (1).
Histopathologic findings of LVNC are unspecific. Myocardial biopsies have been described as normal or with subendocardial fibrosis or fibroelastosis, myocardial fibrosis, myocardial hypertrophy and degeneration, myocardial scarring or signs of inflammation (4, 7, 8). Electron microscopy has shown normal architecture of the myofibrils, with or without mild enlarged mitochondria (7).
Unspecific ECG changes such as left-ventricular hypertrophy, left or right bundle branch block, hemiblock, or ST-T changes may be present (1, 3, 6, 8, 11).
Several differential diagnoses should be considered, such as prominent myocardial trabeculae (typically fewer than three) in a normal ventricle, hypertrophic cardiomyopathy (deep recesses that do not communicate with the ventricular cavity), dilated and restrictive cardiomyopathy, endocardial fibroelastosis, left-ventricular thrombi (particularly in the apex), intraventricular bands, intramyocardial haematomas or abscesses, aneurisms and cardiac metastases (1, 6 – 8, 13).
Genetics and prevalence
Both familial and sporadic cases of LVNC have been described (2, 7, 8, 20, 21). The pathomorphogenetic mechanism behind this congenital heart disease has not been identified, and is probably heterogenous. Several mutations have been found (both in familial and sporadic cases), e.g. chromosome mutations in Xq28 (G4.5-gene), 17p11.2 – 12, 18q12, 11p15, and 5q (CSX -gene) (13, 19). Several of these genes are often affected in other conditions (cardiac and extracardiac) (13, 22, 23). For example, a mutation in the G4.5-gene is also associated with the Barth syndrome, X-linked hereditary endocardial fibroelastosis and X-linked infantile cardiomyopathy (19). Mutations in the MYH7 gene are associated with both LVNC and hypertrophic, restrictive and dilated cardiomyopathies (22). Both X-linked recessive (8, 20) and autosomally dominant (8, 24) inheritance have been described. LVNC presents more often in men than women (6, 8, 9). When symptoms first present in childhood the condition seems to most often be due to X-linked inheritance, whereas adult onset is usually associated with autosomal dominant inheritance (24). The phenotype may vary greatly, even within the same familial hereditary cause (25). It has been noted that facial dysmorphism and other types of dysmorphism are more common with symptom onset in childhood (2, 3). The section for molecular genetics, Medical Genetic Laboratory at Rikshospitalet, Oslo can now perform genetic testing. To our knowledge, routine genetic testing is not recommended in the literature.
Previously, LVNC was described as very rare, but the condition is now diagnosed more often, with various echocardiographic studies showing prevalences in the range 0.05 – 0.24 % (4). The increased occurrence may be due to improved diagnostic possibilities (echocardiography, cardiac MRI) and increased awareness about the disease, but genetic differences between the populations studied may also be an explanation (7). The condition has been diagnosed at all ages, from prenatally up to 92 years (6, 7, 26). LVNC has been identified in 9 % of Australian children with primary cardiomyopathy, and was the third most frequent cause after dilated and hypertrophic cardiomyopathy (27). In a study of adult patients with heart failure, the condition was found in 24 % (28). One possible explanation for the high prevalence in this study may be that the current diagnostic criteria are somewhat unspecific, particularly for individuals of an African genetic origin.
Clinical manifestations and treatment
Clinical manifestations and age at the time of diagnosis vary (2, 5, 6, 8, 16, 29). Although LVNC is a congenital heart disease, cardiac symptoms often present after maturity, and several patients have been described with symptom onset after the age of 60 (3, 7). This may be due to varying degrees of LVNC, and development of concomitant disease, such as ischemic heart disease (1, 30).
Patients may develop progressive heart failure due to systolic and diastolic dysfunction of the LV, with or without right-ventricular dysfunction (1). When LVNC with compromised ventricular function is diagnosed in early childhood, a transient improvement followed by deterioration is often seen (15). Diastolic dysfunction is caused by abnormal relaxation and reduced elasticity of the ventricle. Systolic dysfunction is caused by impaired myocardial contraction in the affected ventricular segments, but poor correlation between oxygen requirements and blood supply to areas with trabecular changes may also be important, and patients with LVNC have typical microvascular dysfunction (1, 13, 31). Heart failure should be treated according to generally accepted guidelines, including heart transplantation if necessary (7, 8, 16, 19, 21, 29, 30).
Several studies have shown an increased risk of serious ventricular tachyarrhythmias and/or sudden death in patients with LVNC (2, 3, 9, 20, 21, 30), whereas others have not revealed any substantially increased risk, even after long observation times (8, 26). Part of the explanation for this discrepancy may be inclusion of patients with more severe disease in the studies first mentioned than in the other two (8). For patients with an episode of potentially serious tachyarrhythmias, there should be a low threshold for implanting a defibrillator (10).
The patients are prone to thromboembolic events (due to thrombus formation in the deep intertrabecular recesses) and supraventricular tachyarrhythmias (2, 3, 8, 9). Prophylactic anticoagulation treatment should therefore be used quite liberally (1, 9, 16).
Various neuromuscular diseases (such as myopathies) are often seen in patients with LVNC (6, 20). In one study, 40 of 49 patients had such a disease (6). Neurological assessment is therefore recommended in patients with this condition (6).
Prognosis
The prognosis is often described as poor, with increased morbidity and mortality due to progressive heart failure, systemic embolization, and malignant ventricular arrhythmias. In one family with Xq28-related LVNC; four children died in infancy, one 8-month-old child developed heart failure, and one child had a heart transplantation at nine months of age (20). A study of eight patients (from 11 months to 22 years of age) reported three deaths after a 5-year follow-up (2). A study with an average observation time of 44 months included 34 patients (with an average age 42 years) at the time of diagnosis; 12 patients died, 18 developed extensive heart failure, eight had thromboembolic events, 14 had episodes of ventricular tachycardia, and four had heart transplants within the study period (9). In a study of 17 patients (age 18 – 71 years), eight died and two had heart transplants within a follow-up period of six years (3). The patients who were asymptomatic at the time of diagnosis had the best prognosis.
However, the question has been raised as to whether the prognosis in general is as serious as indicated above (8, 9, 15, 21, 26, 29). In a Japanese study of 27 patients (age 1 week – 15 years) identified by screening, only two died after a median follow-up of six years, but 14 developed impaired cardiac function in the same observation period (8). A study with an average follow-up of 46 months included 45 patients with an average age of 37 at the time of diagnosis (30 had a dilated and weakened LV and average NYHA class 2,3 at the time of inclusion); only one patient died in the follow-up period (21). Among the patients who had normal left-ventricular systolic function (at the time of inclusion) there were no deaths or heart failures in the observation period. An observational study with an average follow-up period of more than four years included 238 patients (age 1–92 years); only 11 of these had clinical signs or 24-hour ECG findings indicating ventricular tachycardia and only two of them developed potentially life-threatening arrhythmias in the follow-up period (26). A finding of LVNC in an asymptomatic patient, whether the finding is incidental or in conjunction with family screening, is associated with a high likelihood of a stable situation and a good prognosis for many years (29). On the other hand, left-ventricular dilation, symptoms of heart failure, chronic atrial fibrillation, bundle branch block, and previous episodes of potentially life-threatening ventricular tachycardia are noted as obvious negative predictors of survival in these patients (9, 29).
A systematic family screening of 32 asymptomatic relatives showed that six had LVNC and two had echocardiographic left-ventricular anomalies (21). Other studies have also found that these families have an increased prevalence of various cardiac anomalies (22, 23). Due to the uncertain-to-poor prognosis for patients with LVNC, echocardiographic examination of firstdegree relatives is recommended (9, 19).
Conclusion
LVNC has recently been described as a cause of cardiomyopathy. The diagnosis can most easily be made by echocardiography or cardiac MRI. It is important not to overlook LVNC, because it may lead to serious sequelae (such as heart failure, thromboembolic events, ventricular tachyarrhythmias and death), and an early diagnosis may provide these patients with better follow-up and treatment. There is reason to believe that LVNC is more common than previously assumed.
