An elderly man with known myasthenia gravis was admitted as an emergency case after a few days of generally reduced condition, gurgling respirations and shortness of breath. This was the onset of a long and complicated course of illness with pronounced respiratory problems.
The patient was a man in his seventies who developed symptoms of myasthenia gravis in the form of varying ptosis, diplopia and jaw claudication. Blood tests showed an elevated level of acetylcholine receptor antibodies of 6.8 nmol/l (0.0 – 0.4 nmol). He had previously had surgery and radiotherapy for colorectal carcinoma of the Dukes’ B class and had a sigmoidostomy. He was operated on for ventral hernia, complicated by perforation of the small intestine and subsequent peritonitis and wound infection. In addition he had paroxysmal atrial fibrillation and angina pectoris and was being treated for hypertension.
Just over a year after the diagnosis of myasthenia gravis, the patient’s myasthenic symptoms worsened, with weakness in the neck and lower extremities, dysarthria and dyspnoea. The respiratory difficulties could in part be related to chronic obstructive pulmonary disease, which was confirmed during his admission to hospital. Ventilatory support was not required. The symptoms improved following treatment with intravenous immunoglobulin (IVIG), prednisolone and pyridostigmine.
Myasthenia gravis is an autoimmune disease with postsynaptic damage to the neuromuscular junction. This leads to increased fatigue and varying loss of strength in the affected musculature. Acetylcholine receptor antibodies are detected in a large proportion of patients (approx. 90 %). Around half of those remaining have antibodies directed against muscle-specific tyrosine kinase (MuSK).
Due to an enlarged thymus shown by thoracic CT, a thoracoscopic thymectomy was performed after prior plasmapheresis. Histopathological findings revealed WHO type BI thymoma. The procedure was complicated by preoperative bleeding and pneumothorax, making it necessary to switch to an anterolateral thoracotomy. Postoperatively he had pneumothorax, there was air leakage, and he had to undergo reoperation with a wedge resection of the right lung middle lobe and left lung upper lobe. He had postoperative chest pain and was therefore put on gabapentin, a drug which we assumed would not affect his myasthenia gravis. He subsequently returned to good health, was discharged from the neurological ward with no significant symptoms of myasthenia and was prescribed 40 mg × 5 pyridostigmine and 20 mg prednisolone on alternate days.
Two weeks later he was admitted as an emergency case after a few days of reduced general condition, gurgling from his airway and shortness of breath. He had diplopia when looking upwards, but drooping eyelids were not observed. He had dysarthria and increased fatigue of the neck to the point where he only managed to raise his head for a few seconds at a time. Peak expiratory flow (PEF) on admission was only 185 ml/s, and when later the same day this fell to 120 ml/s, he was intubated and connected to a ventilator.
This was the onset of a long and complicated phase in his course of illness, with pronounced respiratory problems.
Treatment with intravenous immunoglobulin was administered at 0.4 g/kg daily for five days. Metoprolol, gabapentin, prednisolone and pyridostigmine were discontinued.
Many drugs are reported to exacerbate myasthenia gravis, among which are metoprolol and gabapentin. Pyridostigmine is used as a symptomatic treatment. The drug can cause cholinergic crisis, particularly at high doses, and thereby further muscle weakness. Additionally increased secretion in the airways can occur as a side effect.
After two days the patient was extubated, but because of significant CO2 retention he had to be reintubated and later tracheostomised. Three days after the first intubation he developed transmural myocardial infarction, with a troponin value of 741 ng/l. He was treated with acetyl salicylic acid, clopidrogel, dalteparin, isosorbidmononitrate and reinstatement of metoprolol.
Since beta blockers may exacerbate myasthenia gravis and the patient required ventilation, metoprolol was discontinued. After thorough consideration the beta blockade was reinstated to aid cardiac function.
Treatment for pneumonia was started simultaneously with cefuroxime, later also with metronidazole, based on raised CRP values, expectoration and possible pulmonary infiltrate visible on chest x-ray. Following immunoglobulin treatment he was still unable to inhale, even with pressure-supported ventilation mode, and needed controlled ventilation. Symptomatic treatment with pyridostigmine was reinstated, and he again received immunosuppressive treatment with 20 mg prednisolone on alternate days.
We chose a conservative initiation of prednisolone, partly in order not to induce fluid accumulation, which in the worst case can result in post-myocardial infarction heart failure, and partly because high doses of glucocorticoids can cause a temporary exacerbation of myasthenia gravis.
When prednisolone was increased to 60 mg daily and pyridostigmine was administered at 60 mg × 5 daily without any notable effect, on day 19 a neostigmine test was performed. Following administration of atropine sulphate, 1.5 mg neostigmine was injected intramuscularly. This acetylcholinesterase inhibitor is most effective after about 20 minutes, and any improvement in the patient’s muscle strength and stamina is assessed. He performed 75 hand contractions without difficulty both before and after the injection. With the ptosis test the eyelid covered the left pupil after 36 seconds, after neostigmine only after 44 seconds, which was not interpreted as a significant improvement. Neither was there any improvement in ventilation ability. Before the neostigmine injection he was able to breathe on his own consistently for four minutes, while half an hour after the injection he only managed to breathe on his own for 90 seconds.
We assessed whether the symptomatic treatment dosage was sufficiently high. The fact that neostigmine did not improve his respiratory capacity indicated that increasing the pyridostigmine dosage would have no better effect and that the first-line treatment should still be immunosuppression.
Methylprednisolone 1 000 mg daily was now administered intravenously for five days, still with no improvement in respiratory function. There were suspicions of pneumonia and the patient was treated with cefuroxime and metronidazole, which was later replaced by piperacillin/tazobactam.
After 33 days of ventilation treatment a new course of IVIG was undertaken over five days. On day 43 we started treatment with azathioprine, increasing to 150 mg a day. The patient was now gradually able to breathe on his own for shorter and later for longer periods, and 44 days after the start of ventilation treatment he breathed on his own for a total of 10 1/2 hours in the course of one day (Figure 1).
After this there was again a significant deterioration when on day 45 he only had autonomous breathing for two hours, the next day for only one hour and afterwards he had no autonomous breathing at all. Since there was a rise in CRP level to 107 mg/l, increased airway mucous secretion and infiltrate at the basal left side on chest x-ray, this was regarded as a further ventilator-associated pneumonia which was treated with clindamycin and ciprofloxacin.
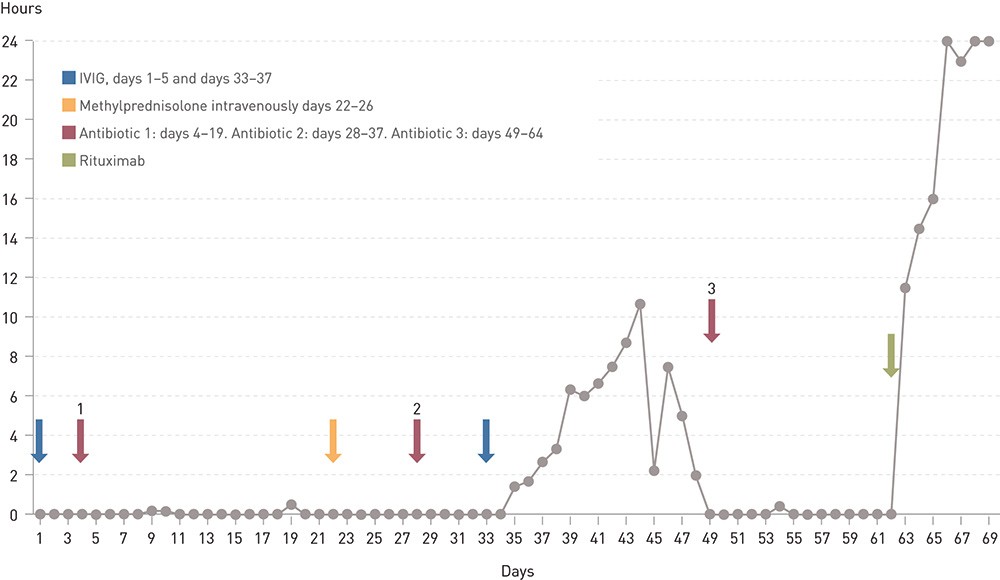
Figure 1 Number of hours per day without respiratory support for a man in his seventies with myasthenic crisis.
The fact that the patient was receiving extensive immunosuppressive treatment while continually contracting infections could later give rise to a dilemma. Even though there was no microbial growth in blood and expectorated sputum cultures, we opted to give antibiotic treatment. The risk of bacterial infection with ventilation treatment is considerable, and infections will sustain the functional impairment in myasthenia gravis. At the same time one must be aware that antibiotics can exacerbate the condition.
After a week the patient was afebrile and free of airway mucous secretion. The CRP level had normalised. However, his respiratory function was still so poor that he was completely dependent on a ventilator. He had signs of dyspnoea when the ventilator was in pressure-support mode.
The ventilation treatment in the hospital’s intensive care unit had now been so prolonged that the possibility of a home ventilator was discussed. In Guidelines for the habilitation and rehabilitation of persons with lung diseases from 2000, myasthenia gravis is indicated as one of the neurological conditions where this can be appropriate. To our knowledge this has not been previously tried in Norway, but it has been used for example in Denmark. Home ventilators are very demanding both financially and in terms of the work required, and their use must be planned and evaluated thoroughly.
The neostigmine test was repeated on day 58, and the ptosis test was now judged to be positive. Before the injection he held his eyelids up for two minutes and 34 seconds, while after the injection they had not yet begun to droop after five minutes. The pyridostigmine dosage was therefore increased from 60 mg × 6 to 90 mg × 6.
When ventilation treatment, two courses of IVIG, intravenous methylprednisolone, peroral prednisolone, azathioprine and pyridostigmine had not improved respiratory capacity after 62 days, it was decided to treat with 1 000 mg rituximab. Respiratory function rapidly improved in the days that followed, and after a short time the patient could be moved to the neurological ward. On day 68 the tracheostomy tube was removed. He could later be discharged home in a good general condition.
Rituximab was again administered after 14 days. On examination six months later the patient had no symptoms or findings to indicate a flare-up of the disease.
Discussion
«Myasthenic crisis» is understood as an exacerbation of myasthenic symptoms to a degree that necessitates intubation, or that makes extubation impossible within 24 hours of a surgical procedure (1). The term is often used more widely to encompass all types of assisted ventilation.
A number of conditions and factors can exacerbate myasthenic symptoms and trigger such a crisis, for example infections, surgical procedures and metabolic disorders. In addition a number of drugs are described as having the potential to cause an exacerbation. Among these are several types of antibiotics, drugs to treat epilepsy, anxiety and pain, and drugs that have cardiovascular effects, such as calcium channel blockers and beta blockers (including eye drops) (2). The list is so extensive that it is almost necessary to take the approach that any drug can exacerbate the condition. The treatment of complications and other simultaneously occurring conditions can therefore be difficult.
In the early 1960s mortality from myasthenic crisis was found to be 42 %, but fell to 6 % at the end of the 1970s (3) and has since remained relatively stable. Over the course of a decade at the Mayo Clinic, USA, 40 patients were treated with a total of 46 cases of myasthenic crisis (4). Extubation failure was defined as the need for reintubation, installation of a tracheostomy or death. There was an extubation failure in almost half of the cases, with reintubation having to be performed nine times, and seven episodes requiring tracheostomy with no attempt at extubation (11 tracheostomies in total). Four patients died. Atelectasis, low pH and low forced vital capacity (FVC) upon extubation were the main predictors for reintubation.
Myasthenia gravis may affect different muscle groups to very varying degrees. Muscles connected to the eyelids and eye movements may be affected without any involvement of other muscles (ocular myasthenia), but other types of focal affection are also known. Selective affection of the laryngeal muscles with vocal cord paresis and stridor has been reported (5). In dogs, which have a striated musculature in the oesophagus, an affection of this musculature with acetylcholine receptor-positive myasthenia gravis has been described without other weakness being recorded (6). This leads to oesophageal dilatation and reflux.
Muscle-specific tyrosine kinase (MuSK) is important in the formation and maintenance of neuromuscular junctions. It has been shown in mice that the expression and level of this protein varies between different muscles. Different levels determine the ability to form ectopic clusters of acetylcholine receptors under certain conditions (7). It may appear plausible that different expressions of proteins in different muscle cells result in different plasticity and vulnerability. After a short time our patient had strength and stamina in the muscles of the extremities, but was nevertheless completely dependent on ventilation because of pronounced affection of the respiratory muscles.
The personnel treating the patient tended towards favouring different approaches to the treatment. The anaesthesiologists wanted early respiration training to wean the patient off ventilatory support, while the neurologists wanted to relieve the respiratory muscles until the receptor apparatus had an opportunity to regenerate itself. Sensible training cannot be ruled out as beneficial to prevent degeneration of the respiratory muscles from prolonged ventilation treatment (8, 9). However, there is no doubt that as long as the neuromuscular junction of the respiratory muscles is not functioning, the patient will be unable to breathe autonomously.
In treating myasthenia gravis one seeks to optimise the transfer of signals in the neuromuscular junctions. Administration of an acetylcholinesterase inhibitor, normally pyridostigmine, will increase the amount of transmitter substance and strengthen the effect on the receptors. Pyridostigmine is usually well tolerated, but can give side effects such as nausea, diarrhoea, increased mucous secretion in the airways, bradycardia and atrioventricular block (AV block). At high doses the acetylcholine receptors can become de-sensitised, which will result in muscle weakness, i.e. cholinergic crisis.
The treatment is also directed against pathogenic antibodies in order to reduce postsynaptic damage. Corticosteroids have an immunosuppressive effect which in most cases improves the course of illness. However, the side effects accompanying such treatment are not insignificant and often induce weight-gain, diabetes and osteoporosis. Azathioprine is therefore often used for steroid-sparing immunosuppression. Other alternatives might be cyclosporine, cyclophosphamide or methotrexate. The effectiveness of extirpation of the thymus in the elderly is uncertain, but thymectomy is always indicated for thymoma (10).
In myasthenic crisis the immunomodulating treatment is intensified with intravenous immunoglobulin or plasmapheresis. High-dose intravenous corticosteroid therapy may also be appropriate. The elimination of any triggering factors must always be sought.
Different levels of acetylcholine receptor antibodies among different patients do not always correspond to the degree of severity of the disease, but changes over time in the same person may indicate a change in disease activity. At the time of diagnosis our patient had an antibody level of 6.8 nmol/l. After a course of 55 days of the myasthenic crisis, the level was ten times as high, 68.0 nmol/l. Two days later the level was 59.0 nmol/l.
There are many factors that may have finally contributed to the improvement in our patient’s respiratory capacity. Several courses of antibiotics were given, and the infections were fought. The intensified symptomatic treatment with pyridostigmine may have had some significance but was not decisive, and the improvement came several days after the last increase in dosage. The main reason was surely the prolonged and finally comprehensive immunosuppressive treatment. At extubation the patient was on high-dose prednisolone, azathioprine was introduced and two courses of IVIG were undertaken. Rituximab was given six days before extubation.
Rituximab is a monoclonal antibody directed specifically against CD20-positive B cells and is used for lymphoma, chronic lymphocytic leukaemia and severe rheumatological diseases. There are anecdotal reports of a beneficial effect also in myasthenia gravis (11). Five patients at Sahlgrenska Hospital are described with severe myasthenia gravis who received this treatment. All showed marked improvement, but it was slow and gradual (12). The improvement in our patient was much more rapid, and it is uncertain how much can be attributed to rituximab. A case history has also been published in which a 31-year-old woman with refractory myasthenia gravis and respiratory failure was treated for several months with a total of three courses of intravenous immunoglobulin, prednisolone, cyclophosphamide and pyridostigmine, without lasting effect. After treatment with rituximab there was a rapid improvement in respiratory function; however the time period is not precisely indicated (13).
Conclusion
The course of illness described shows that the loss of function with myasthenic crisis can be very prolonged. It is important to remember that in the great majority of cases the condition is reversible. No controlled studies exist regarding rituximab for myasthenia gravis, but there are reasons to believe that the drug may have a beneficial effect in severe cases.
