Hereditary haemochromatosis is a congenital disorder which affects the regulation of iron metabolism thus causing increased gut absorption of iron and a gradual build-up of pathologic iron deposits in the liver and other internal organs, joint capsules and the skin (1). From the age of 40 – 50 the iron overload may cause serious disease, which in its final stages is characterised by liver cirrhosis, diabetes and bronze-coloured skin, also referred to as «bronze diabetes». The prevalence in the ethnic Norwegian population of homozygous and heterozygous inheritance is 0.8 % and 12 – 15 % respectively, which makes haemochromatosis one of the most common hereditary diseases in Norway (2).
This article describes a development which over the course of 150 years has transformed haemochromatosis from a life-threatening disease into a disorder which can be picked up in its preclinical stage by routine blood tests and which allows the prevention of disease by venesection therapy. This is probably the only course of treatment dating back to Antiquity which still has a place in modern medicine (fig. 1, fig. 2) (3).
Figure 1 Venesection. Mediaeval illustration. British Library, London
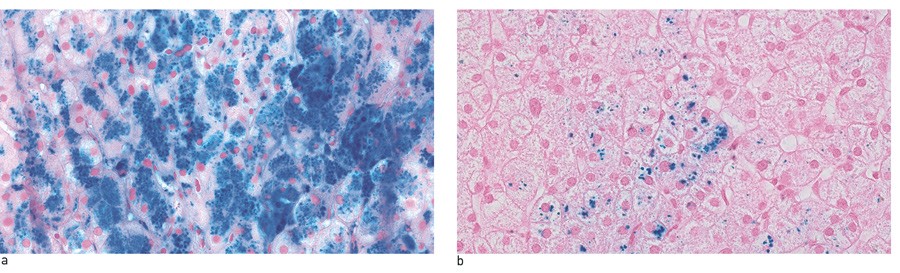
Figure 2 Liver biopsy from a man in his 50s with hereditary HFE haemochromatosis and homozygous for the C282Y mutation, a) before and b) after 38 venesections over the course of 12 months. At the time of diagnosis, his serum ferritin was 3780 μg/l and his transferrin saturation 98 %. In total, ca. 18 litres of blood were extracted, and 8 – 10 g iron removed. After treatment, his serum ferritin was 72 μg/l and the transferrin saturation 40 %. The patient has consented to the pictures being published.
The pioneers
The first description of a patient with haemochromatosis has been attributed to a French physician, dr. Armand Trousseau (1801 – 67), who in 1865 gave a lecture on glycosuria and a new syndrome presenting with diabetes, liver cirrhosis with yellow-grey granules and brown skin pigmentation in a 36-year-old male (fig 3) (4).
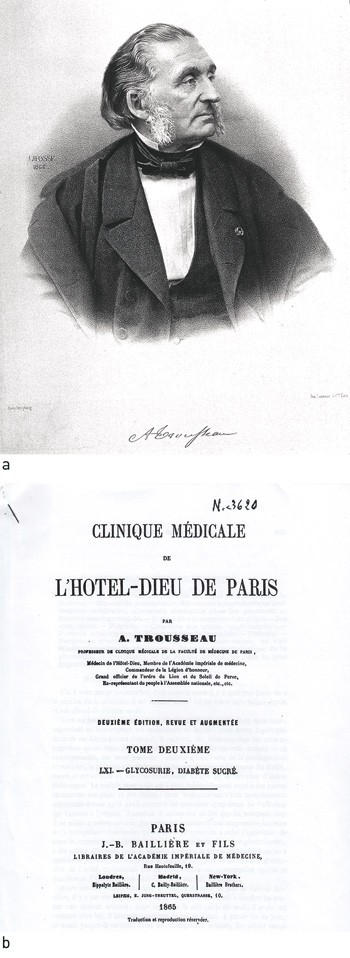
Figure 3 a) Armand Trousseau. Lithography by J.B.A. Lafosse from 1866 (CC BY 4.0). b) First page of article from 1865 in which Trousseau describes the first known case of haemochromatosis. Clinique médicale de l’Hôtel-Dieu de Paris, digitised by the University of Ottawa.
The second case report was published in 1871 by his compatriot, professor Charles Troisier (1844 – 1919) (5). At the time, it was not known that the pigment was in fact iron, and the disease was referred to as «pigment cirrhosis» until professor Victor Hanot (1844 – 96) introduced the name of «bronze diabetes» in 1886 (6).
Rudolf Virchow (1821 – 1902), in his autopsies on organs with internal bleeding, had described a yellow-brown pigment referred to as haematin as early as in 1847 (7). The pigment could be stained Berlin blue and thus contained iron. Berlin blue was a staining method known from Berlin, where alchemists in 1704 had discovered that a combination of blood, iron sulphate and potassium carbonate formed an insoluble dark blue pigment (8). The pigment was also called Prussian blue because it was used to dye the uniforms of the Prussian army. In 1867 the German pathologist Max Perls (1843 – 81) improved the staining method by using potassium ferrocyanide which reacts specifically with trivalent iron oxide (9). Perls’ staining reaction became the standard histological technique used to demonstrate iron pigment, which since 1888 has been called «haemosiderin» (10).
In 1889 the German doctor Friedrich von Recklinghausen (1833 – 1910) was the first to stain liver autopsies from patients with bronze diabetes, thus demonstrating that the yellowish brown pigment was caused by massive iron deposition (11). He believed that the iron stemmed from internal bleeding with haemolysis and catabolism of haemoglobin, which is why he named the pigment «haemochromatosis».
Over the next few decades, discussions about the cause of haemochromatosis centred around two main theories (12). The first theory argued that haemochromatosis was secondary to diabetes, but this idea was gradually dropped as it became clear that most patients with diabetes did not suffer from iron overload. However, patients suffering from haemochromatosis did have diabetes and haemosiderin deposits in the pancreas, which is why «bronze diabetes» came to signify late-stage haemochromatosis.
The second theory argued that portal liver cirrhosis was the primary cause, a stance explained by cirrhosis being a consistent post mortem finding in cases of haemochromatosis. However, in the early 1930s it was established that liver cirrhosis is rarely accompanied by haemochromatosis. Nevertheless, a link was not ruled out, and some predicted the true answer by maintaining that iron deposits in the liver could cause sclerosis, and that the iron stemmed from food.
At the same time, the haemolysis theory was discarded – a theory on the origin of iron which had ruled supreme since the time of von Recklinghausen. It was now scrapped because it was impossible to demonstrate that internal bleeding or any other haematological change was the cause of the haemosiderin deposits in the liver (12). Theories of infection, toxins and chronic lead and copper poisoning could not be verified. The answer to where the iron came from remained illusive, and the disease came to be known as «idiopathic haemochromatosis».
Joseph Sheldon
In 1935 the English doctor Joseph Sheldon (1894 – 1972) published Haemochromatosis, a retrospective review of all patients in the medical literature who so far had been diagnosed with haemochromatosis (12). Of 311 patients, 295 were male and 16 female; 63 % were over 45 years of age and life expectancy after the clinical diagnosis was approximately 18 months. In most cases, the diagnosis had been confirmed post mortem by histological examination of autopsies of all the body’s organs and tissues. Iron was found in all organs apart from the central nervous system. The book came to be a standard work which to this day provides the most complete picture of iron deposition in the body in cases of late-stage haemochromatosis.
Sheldon introduced a new theory, that haemochromatosis might be a congenital metabolic disorder involving a molecular defect which inhibited iron export from cells, and that retention of iron led to secondary cell damage and disease. The fact that a congenital disease was not discovered until mid life, indicated that the iron overload would have to be substantial in order to cause organ damage. In his opinion, small daily iron deposits of up to 2 mg would explain the clinical end stage with an iron overload of 30 – 40 g at the age of 45 – 50.
The theory was clinically logical, but incorrect in terms of explaining the accumulation of iron in cells, and it shed no light on the origin of the excess iron. Sheldon ruled out alcohol as a cause of the disease, but the alcohol theory had considerable support from others, and as late as in 1963 it was purported that haemochromatosis was an acquired disease caused by high alcohol consumption (13).
Sheldon ended his book with a prophesy that haemochromatosis was the key to understanding how cells normally process metals, which at the time was «shrouded in mystery». He was proven right: after the discovery of the haemochromatosis gene in 1996 the main elements of the molecular regulation of the body’s iron balance were quickly described.
The discovery of ferritin and transferrin
The discovery of the iron-binding proteins ferritin and transferrin were milestones in the development of modern diagnostics and the treatment of haemochromatosis and other diseases of iron metabolism.
First off the mark was the Czech professor Vilém Laufberger (1890 – 1986), who in 1937 successfully crystallised a large iron-binding protein from horse spleen (14). He called the protein «ferritin», in honour of the German pharmacologist Oswald Schmiedeberg (1838 – 1921), who as early as in 1894 had isolated a ferrous protein compound from swine liver which he referred to as ferratin (Latin: ferratus, bound to iron) (15). It was soon demonstrated that ferritin in the liver, spleen and bone marrow functioned as an iron store (16, 17). Molecular studies took a little longer, but in the early 1970s the structure of ferritin, and its function as an intracellular iron store, was mapped in detail (18). Haemosiderin was formed by the catabolism of ferritin in the lysosomes (19).
To start with, ferritin could only be measured in serum after acute liver disease with cell necrosis (20), but after the development of a sensitive immunological assay in 1972, it became possible to measure ferritin in serum from healthy individuals, and it was demonstrated that the level was proportional to the volume of the iron deposits in the liver and bone marrow (21).
The analysis of serum ferritin in clinical work was a major step forward which improved the diagnostic process for iron deficiency and enabled the discovery of iron overload long before it had grown to a scale sufficient to cause organ damage. For people with a congenital predisposition to haemochromatosis, the possibility of a diagnosis at the preclinical stage, using serum ferritin, brought radical improvement of their prognosis (22).
In 1947, ten years after the discovery of ferritin, transferrin was isolated from swine serum and characterised by the Swedish doctor Carl-Bertil Laurell (1919 – 2001) and his colleagues (23). This discovery formed the climax of a lengthy search for a plasma protein that was capable of carrying iron to the cells, following the demonstration in 1925 of non-haemoglobin-bound iron in plasma. Cases of haemochromatosis had previously shown high concentrations of iron in serum, and high transferrin saturation between 50 and 100 % now became a criterion for making the diagnosis (24).
By the early 1950s more than 1,000 scientific articles in the literature had established the basics of our current knowledge about serum iron and the biochemistry, physiology and clinical significance of transferrin.
In the 1970s a biochemical serum pattern with stable elevated ferritin and high transferrin saturation became pathognomonic of haemochromatosis, and iron staining of liver biopsies ceased to form a routine part of the diagnostic process.
Pathophysiology
The pathophysiology behind haemochromatosis was deduced from balance studies of iron absorption and excretion. The breakthrough came after Robert McCance (1898 – 1993) and Elsie Widdowson (1906 – 2000) had pointed out in 1937 that missing evidence of excretion of iron in the intestines and kidneys was an indication of the iron balance being regulated by the absorption of iron (25). This theory was later verified by numerous studies, culminating in 1968 with a large multicentre study which demonstrated that the iron loss in men was stable and very low, only ca. 1 mg per day, and that it was only a matter of passive loss of iron in cells that were rejected by the intestinal mucous membrane, urinary tract, bile duct and skin (26). The same applied for haemochromatosis.
Sheldon’s hypothesis of «an inborn error of metabolism» would therefore have to apply to the enterocytes, which absorbed more iron than necessary so that net iron absorption exceeded the physiological demand. The effort to find the defect was escalated, but the results were nonetheless illusive. The closest they came was to suggest that the normal mechanism for preventing excessive iron absorption – launched in 1943 as a form of «mucosal block» – was missing in cases of haemochromatosis (27).
Sixty years later, in the early 2000s, it was established that the block theory was generally correct, but that the true explanation was very different from earlier speculations.
Genetics and molecular regulation
The turning point that verified the hereditary theory came in 1976, when an excess of tissue antigens HLA-A3 and -B14 was established in families affected by haemochromatosis (28). The search for a «haemochromatosis gene» in the vicinity of the HLA-locus on chromosome 6, now became more focused, and the illness which had previously been termed «idiopathic» was now renamed «hereditary» haemochromatosis.
The 1990s’ revolutionary developments in gene research, which gave access to new genetic analysis techniques, were a precondition for the breakthroughs in the mapping of molecular iron metabolism. This started in 1996 with the discovery of two new point mutations in the human genome, of which the most important, C282Y, was found in 85 % of patients suffering from haemochromatosis, while the other – H63D – had less of a clinical effect (29). The new gene (and protein) was referred to as HFE and the term «HFE haemochromatosis» came to be commonly recognised. The C282Y mutation is assumed to have arisen in Europe more than 4,000 years ago (30). Its geographical distribution coincides with Viking travel routes, which suggests that the mutation spread with the Vikings. This explains why the vernacular name for haemochromatosis used to be «the Viking disease» (fig. 4) (31).
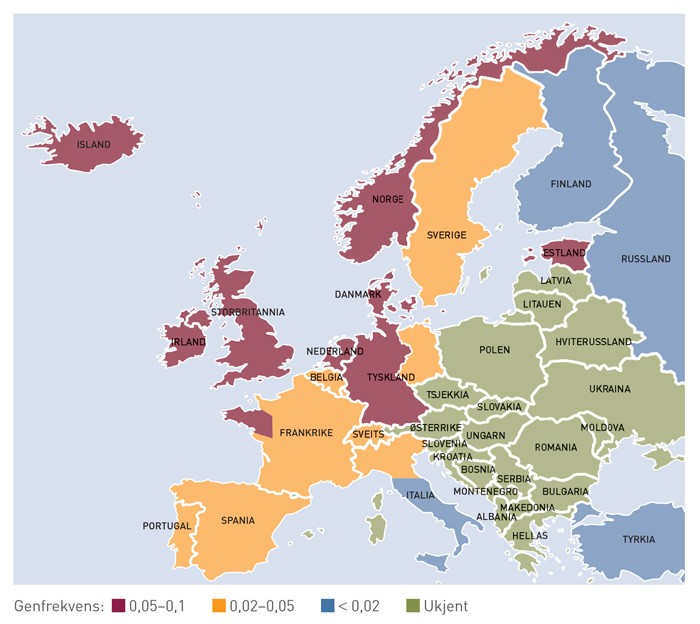
Figure 4 Map showing the spread in Europe of the most important haemochromatosis mutation, C282Y. The highest prevalence is found in the countries around the North Sea, where the Vikings were marauding almost 1,000 years ago. The map is slightly misleading with respect to Norway, as the stated frequency is probably somewhat lower in Northern Norway. (31)
The function of the newly discovered HFE protein became clear after the breakthrough discovery in 2000 of the hormone peptide hepcidin in plasma and the cell membrane protein ferroportin. Ferroportin, which is the only protein known to be able to export iron from cells, reacts with hepcidin which slows down the iron export (32). HFE, interacting with other genes, regulates the synthesis of hepcidin in the liver and therefore also the plasma concentration of hepcidin, so that the import of iron to plasma is controlled by physiological demand. In the case of iron overload, HFE is activated so that the plasma concentration of hepcidin increases and gut absorption of iron is reduced. Iron deficiency on the other hand, leads to deactivation of HFE so that the rate of hepcidin synthesis is reduced and the gut absorption of iron increases. In cases of hereditary haemochromatosis with loss of HFE function, the rate of hepcidin synthesis is permanently reduced and the iron absorption elevated – despite a growing iron overload. Hepcidin is the most important regulator of the iron balance, and reduced rates of synthesis and plasma concentration are common denominators for the HFE mutation as well as rare mutations in other contributory genes that cause haemochromatosis.
Venesection therapy
The idea of treating haemochromatosis with intermittent venesection was first attempted on a patient in New York in 1941 in the hope that, by stimulating blood production, the excess iron would be used to increase the rate of haemoglobin synthesis and thereby disappear (33). The treatment was never completed and attracted no attention. A few years later another US hospital performed venesection on five patients, one of whom over the course of 16 months had 40 litres of blood extracted, with a total of 20 grams of iron removed (34). The authors were among the first to make use of liver biopsy for diagnostic purposes and in order to check the efficacy of the venesection. The liver was emptied of haemosiderin and the cirrhosis showed signs of improvement. The patients felt better subjectively and they contracted neither anaemia or any other side effects.
The introduction of intensive venesection therapy in the early 1950s gave a better quality of life and extended the life expectancy of seriously ill patients suffering from hepatomegaly and diabetes (35). Preventive venesection at the preclinical stage of haemochromatosis resulted in a normal lifespan (36).
Conclusion
Haemochromatosis was originally a clinical diagnosis in patients suffering from serious illness. Thanks to our current ability to discover iron overload at the preclinical stage by means of routine blood tests, haemochromatosis has been transformed from a life-threatening illness to a risk factor which can be kept in check by preventive venesection therapy.
Mapping of molecular iron metabolism and its importance for the iron balance gives us hope that we may find new forms of treatment for haemochromatosis. For the time being, the development of a drug with a hepcidin-like effect is the most promising alternative (37).
