Immunoglobulin G subclass 4-related disease (IgG4-RD) is a relatively recently identified immune-mediated systemic disease characterised by inflammation and progressive fibrosis. The most common clinical manifestations are pancreatitis and sialadenitis, but the disease may affect virtually any organ in the body. Microscopic examination of biopsy specimens reveals inflammation with a substantial proportion of IgG4-positive plasma cells and a characteristic fibrosis pattern. Glucocorticoids are the first-line choice of treatment.
IgG4-related disease is a rare and relatively recently identified condition that has been viewed since 2003 as an immune-mediated systemic disease. The disease may affect virtually any organ or structure in the body, and can in principle be compared with sarcoidosis: an inflammatory condition with heterogeneous clinical presentations demonstrating characteristic histopathological findings in affected organs.
Microscopy of biopsy tissue reveals lymphoplasmacytic infiltrates and a characteristic circular fibrosis pattern called storiform fibrosis (fig. 1a). A large proportion of the infiltrating plasma cells express B-cell receptors of the IgG4 subclass (IgG4-positive plasma cells), which has given the disease its name. Massive infiltration of immune cells into parenchyma and veins in affected organs may lead to tumour formation and obliterative phlebitis, respectively (1).

Figure 1 Histology specimens from patient with IgG4-related disease with pancreas affection. a) Section of pancreas. Haematoxylin and eosin stain (HE). 20x enlargement. Storiform fibrosis and lymphoplasmacytic inflammation surrounding residual preserved acinar pancreatic tissue, fatty tissue and some outlet ducts. b) Pancreas. HE. 80x enlargement. Outlet ducts with pronounced subepithelial lymphoplasmacytic inflammation. c) Pancreas. Immunohistochemical staining for IgG. 80x enlargement. The majority of the subepithelial plasma cells are IgG-positive (brown colour). d) Pancreas. Same section as (c). Immunohistochemical staining for IgG4. 80x enlargement. Almost all the IgG-positive plasma cells are also positive for IgG4 (brown colour)
The epidemiology has been only partly determined, but prevalence is estimated to be 0.28 – 1.08 cases per 100 000 (2). The average age at the time of diagnosis is 60 years. Men are affected more often than women, particularly with pancreatobiliary involvement, where the gender ratio is 3:1. Gender differences are less pronounced among patients with salivary gland involvement (1). The reason for gender differences in organ involvement is not yet known. The condition is described in a case report in Tidsskriftet (3).
Background
In 2001, Hamano et al. found serum IgG4 levels in 20 patients with autoimmune pancreatitis that were elevated compared with the control group (4). Two years later, the same patient group was found to have lymphoplasmacytic inflammation with high levels of IgG4-positive plasma cells in pancreas biopsies, and extra-pancreatic lesions with a similar histopathological and immunohistochemical phenotype (5). This gave rise to the hypothesis of an underlying systemic disease with multi-organ involvement. Similar findings have subsequently been observed in virtually all organs and structures, including bile ducts, aorta, meninges, salivary glands, orbits, lungs, kidneys, retroperitoneum and lymph nodes.
The concept of IgG4-related disease was introduced in 2011 (6), and now encompasses a number of conditions previously regarded as isolated, idiopathic entities, including autoimmune pancreatitis, Mikulicz’s syndrome (sclerosing sialodacryoadenitis), Riedel’s thyroiditis and Ormond’s disease (idiopathic retroperitoneal fibrosis (box 1). These conditions represent different manifestations of the same fibroinflammatory systemic disease: IgG4-related disease.
Box 1 Some conditions that are now regarded as part of IgG4-related disease. Adapted from Kamisawa et al, 2015 (1)
Immunoglobulins
Plasma cells are activated B-lymphocytes and produce and release immunoglobulins (antibodies). An immunoglobulin monomer consists of two heavy and two light chains that together form two fragment antigen-binding (Fab) regions and one fragment crystallisable (Fc) region (fig. 2).
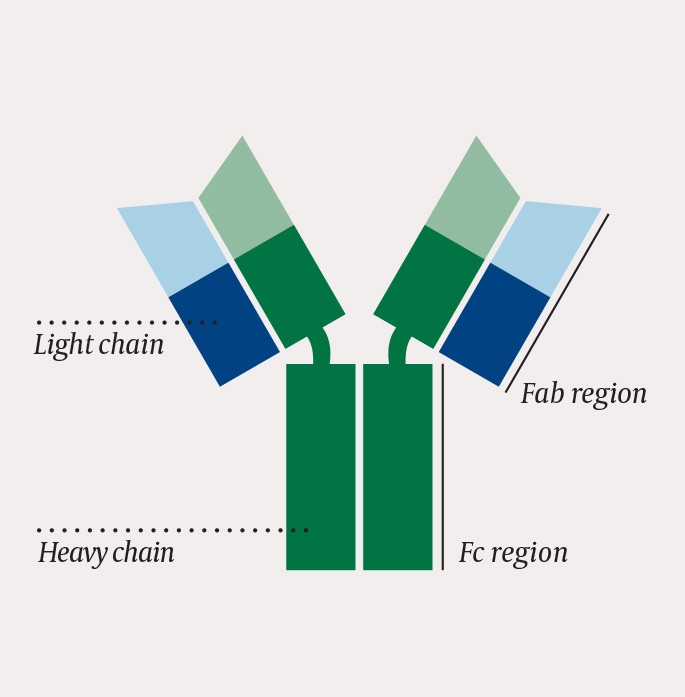
Figure 2 One immunoglobulin monomer consists of two heavy and two light chains that together form two Fab regions and one Fc region
The Fab regions bind antigen and determine the affinity and specificity of the immunoglobulin. All immunoglobulins produced by a single plasma cell have identical Fab regions and recognise the same antigen.
The Fc region dictates the immunoglobulin class and function. There are five main classes of immunoglobulins: IgM, IgD, IgG, IgA and IgE. The Fc region activates the immune system’s effector systems, such as macrophages, neutrophils and complement. The five classes have structurally different Fc regions with differing ability to activate various effector systems and produce different immune responses.
Which class is optimal depends on the type of antigen stimulus to which the body is subjected. For example, IgG will effectively combat extracellular bacteria, while IgE contributes to a potent defence against parasites. Surrounding activated immune cells, such as T-lymphocytes, produce signalling molecules (cytokines) which by means of paracrine signalling stimulate plasma cells to change Fc region (for example, from IgM to IgG) to the class that is most effective in combating the pathogen in question. Since this change only takes place in the Fc region, a class change of this nature will not affect the Fab regions. The immunoglobulin continues to recognise the same antigen, but the effect of the antigen binding is optimised so that the most advantageous effector systems are activated.
Immunoglobulin G4
There are four subclasses of immunoglobulin G, IgG1, IgG2, IgG3 and IgG4. These subclasses have Fc regions that are almost identical, but small differences in their primary structure give the subclasses different functions. IgG1 is the dominant subclass in serum, while IgG4 only constitutes about 5 % of the total IgG concentration in healthy individuals (7). Elevated serum IgG4 levels are seen in about 60 % of patients with IgG4-related disease. This is a non-specific finding, as elevated serum IgG4 is also found with other conditions (7, 8).
IgG1 is a proinflammatory immunoglobulin that induces potent activation of macrophages and complement. This is also true, to varying degrees, of IgG2 and IgG3. IgG4 does not have the same property of activating effector systems, and therefore has a limited proinflammatory effect. IgG4 can also bind to, and neutralise, the Fc region of other IgG subclasses (7). IgG4 therefore has a net anti-inflammatory effect, which may seem paradoxical given that IgG-related disease is characterised by inflammation. This has challenged the theory of the role of IgG4 in the pathogenesis of the disease.
One theory is that the high levels of IgG4-positive plasma cells are a consequence, and not a cause, of the inflammation associated with IgG4-related disease (1). According to this theory, the tissue destruction is due to persistent activation of CD4-positive T-lymphocytes, which in turn activate fibroblasts and macrophages, resulting in fibrosis formation. It is further postulated that release of cytokines by activated T-lymphocytes stimulates local plasma cells to undergo a change of class to IgG4. According to this hypothesis, the IgG4 predominance is therefore secondary to the underlying pathological process. IgG4-related disease is a hyperinflammatory condition, and it seems plausible that IgG4 is not directly causative in view of the protein’s lack of proinflammatory properties. This theory is challenged by the fact that the monoclonal anti-CD20 antibody rituximab appears to be an effective treatment for IgG4-related disease. Rituximab acts through antibody-, cell- and complement-dependent mechanisms to cause selective depletion of B-lymphocytes, as these are the only cells that express the CD20 protein in the cell membrane. This results in reduced plasma cell levels and hence reduced production of immunoglobulins, mainly IgG and IgM. Why, then, does B-lymphocyte depletion have a therapeutic effect, if the disease is mediated by T-lymphocytes, and B-lymphocytes and IgG4 are not involved in the pathogenesis? One possible explanation is that it is antigen-presenting B-lymphocytes that mediate the persistent activation of the T-lymphocytes, and that depletion of these B-lymphocytes brings the pathological T-lymphocyte response to a halt (9). This is consistent with the theory that the immunoglobulins are only passive participants in the pathogenesis of the IgG4-related disease.
Other theories concerning the pathogenesis of IgG4-related disease indicate mast cells, basophils, eosinophils and plasmablasts as influential participants. Activation of macrophages and complement has also been postulated as a central mechanism (8). As with other chronic inflammatory conditions, it is plausible that an environmental factor such as infection in genetically predisposed individuals results in loss of immunological tolerance, with a consequent persistent immune response (1).
Genetic factors have only been partially mapped, but candidate genes have been identified that may be associated with the development of IgG4-related disease with pancreatic involvement, including the HLA alleles DRB1*0405 and DRB1*0401, and polymorphisms in the genes FCRL3 and CTLA4, which are involved in regulating B- and T- lymphocytes, respectively (7, 10).
The pathogenesis of IgG4-related disease is probably multifactorial and complex, and has only been partly determined.
Workup
IgG4-related disease is diagnosed on the basis of clinical, biochemical and histopathological findings. It is essential to exclude differential diagnoses, some of which are shown in box 2. In 2012, an international multi-disciplinary expert group introduced diagnostic criteria (11) whereby the number of IgG4-positive plasma cells per field of view (x40) in combination with lymphoplasmacytic inflammation, storiform fibrosis and/or obliterative phlebitis in the histological sample are used to assess the probability of IgG4-related disease. Several organ-specific diagnostic criteria have also been proposed.
Box 2 Some relevant differential diagnoses
Malignancy (lymphoma, sarcoma, cholangiocarcinoma, pancreatic cancer, etc.)
Biochemical analyses
Elevated serum IgG4 concentration is seen in the majority of patients, but this is neither a sensitive nor a specific finding, and must be interpreted with caution. An elevated serum IgG4/total IgG ratio is indicated to have somewhat higher specificity (1). The serum level of plasmablasts (a stage between B-lymphocytes and differentiated plasma cells) is often high and appears to have greater diagnostic value than serum IgG4, but at the present time this test is not available for clinical use (7). Clinical experience shows that patients may have a persistently high C-reactive protein and sedimentation rate, but these may also be in the normal range (7, 12).
Histology and immunohistochemistry
Histopathological examination shows lymphoplasmacytic inflammation, and immunohistochemical examination reveals a substantial percentage of IgG4-positive plasma cells (fig. 1 a–d). The number of IgG4-positive plasma cells per field of vision (40x) required to consider the diagnosis depends on the organ under investigation. An IgG4/IgG-positive plasma cell ratio greater than 0.4 further increases the probability of IgG4-related disease (11). Storiform fibrosis and obliterative phlebitis, where infiltrating plasma cells obliterate the vein lumen, are other typical findings.
Diagnostic imaging
Diagnostic imaging is not capable of distinguishing IgG4-related disease from differential diagnoses such as malignancy. A PET scan may be of value for assessing the extent of disease and response to therapy (1).
Clinical phenotypes
IgG4-related disease is regarded as a single systemic disease with variable organ involvement.
Symptoms develop gradually secondarily to expansive and/or occluding processes, such as exophthalmos or obstructive icterus. Non-specific symptoms such as abdominal pain, weight loss and fatigue occur (1, 13). In a Japanese cohort study with 235 patients with IgG4-related disease, pancreatitis was the most common manifestation (60 %), followed by sialadenitis, tubulointerstitial nephritis, dacryoadenitis and periaortitis (13). Over half the patients had two or more affected organs.
Autoimmune pancreatitis
There are two types of autoimmune pancreatitis: type 1 (lymphoplasmacytic sclerosing pancreatitis) and type 2 (idiopathic duct-centric pancreatitis). Only type 1 is regarded as a manifestation of IgG4-related disease. The typical presentation is abdominal pain, possibly obstructive icterus secondary to parenchymal fibrosis or accompanying IgG4-related sclerosing cholangitis.
Mikulicz’s syndrome
Sclerosing sialodacryoadenitis is characterised by painless enlargement of the lacrimal, parotid and/or submandibular glands, with or without sicca phenomena. In contrast to Sjögren’s syndrome, male predominance and the absence of anti-SSA and anti-SSB autoantibodies are seen, and a clinical response to corticosteroids.
Tubulointerstitial nephritis
Tubulointerstitial nephritis is the most common intrarenal manifestation of IgG4-related disease, but is often asymptomatic and detected through targeted testing of patients with extrarenal IgG4-related disease (7). Imaging may reveal renomegaly and/or low-attenuation renal lesions. In symptomatic patients, the clinical findings vary from microalbuminuria to acute renal failure. In contrast to drug-induced tubulointerstitial nephritis, patients with IgG4-related disease do not normally have fever, CRP elevation, pyuria or white blood cell casts in the urine (12).
Retroperitoneal fibrosis and periaortitis
Retroperitoneal organs such as ureters and aorta may be affected, leading to hydronephrosis and periaortitis, respectively. This was previously known as Osmond’s disease (idiopathic retroperitoneal fibrosis). Other causes of retroperitoneal fibrosis such as drugs, infections and malignancy must be excluded (7).
Other manifestations
Patients often have localised or generalised painless lymphadenopathy. Fever is rarely seen. Exophthalmos may be a result of dacryoadenitis or orbital pseudotumours (1). Pituitary involvement may cause hormone disorders and expansive complications such as headache and bitemporal hemianopsia (7). Cerebral affection is unusual, but diffuse thickening of the meninges (hypertrophic pachymeningitis) has been described and may give rise to symptoms such as headache, radiculopathy or cranial nerve palsies (7).
Who should be assessed?
Indication for assessment for IgG4-related disease is determined on an individual basis, preferably in consultation with a rheumatologist. Assessment should be considered for patients with the following clinical presentations: pancreatitis with no obvious cause, sclerosing cholangitis, bilateral enlargement of the salivary or lacrimal glands, retroperitoneal fibrosis, orbital pseudotumours and/or exophthalmos.
Treatment
The first-line choice for treatment for IgG4-related disease is high-dose peroral prednisolone (0.6 mg/kg) for 2 – 4 weeks, followed by gradual tapering to a maintenance dose of 2.5 to 5 mg daily. The duration of maintenance treatment has not been defined. Most patients have a prompt clinical response to corticosteroids (7), but recurrence in connection with tapering or cessation is frequently seen. Corticosteroids reduce inflammation and may bring about an improvement in tissue function. Early treatment is crucial for avoiding progressive fibrosis with irreversible loss of organ function. The natural course in untreated patients has not been established.
There are no prospective studies of the use of traditional steroid-sparing drugs such as methotrexate and azatioprine. Rituximab has been used, as mentioned, but randomised studies are required to establish an optimal treatment strategy.
Summary
IgG4-related disease is a rare, immune-mediated systemic disease that causes inflammation and fibrosis in affected organs. Pancreaticobiliary involvement is the most common, but over half of all patients have multi-organ involvement. Because organ involvement is variable, the disease is a relevant differential diagnosis for a number of clinical presentations, and virtually all subspecialists may encounter these patients in their practice. Early treatment is crucial for avoiding progressive fibrosis and irreversible loss of organ function.
