Fever, cough and elevated inflammatory markers are suggestive of pneumonia. But when ascites and striking radiological findings from multiple organ systems are also present, a number of differential diagnoses should be considered. A broad diagnostic examination may be required to make the correct diagnosis, which in this case turned out to be a rare and little known condition.
A woman in her sixties contacted her doctor with symptoms of deep vein thrombosis (DVT) following a fracture of her left ankle. She had a history of glaucoma, hypertension and hypercholesterolaemia. Deep vein thrombosis was confirmed by ultrasound examination, and she was also found to have thrombocythaemia with a platelet count of 1026 · 109/l (145–390). In addition, she had leukocytosis with a leukocyte count of 12.3 · 109/l (3.7–10.0), while her haemoglobin level was 13.8 g/100 ml (11.7–15.3). Further testing revealed a JAK2-V617F mutation, and essential thrombocythaemia was diagnosed. Treatment with hydroxyurea and anticoagulants was initiated in accordance with relevant guidelines.
Despite treatment, the patient had persistent swelling of her left lower limb, and CT venography was therefore performed. As well as oedema extending up to the thigh, the examination revealed pronounced oedema in intraabdominal adipose tissue, swollen lymph nodes in the groin and mesentery, and reactive changes around several abdominal organs, including the kidneys. The subsequent workup included several radiological examinations, which did not help clarify the diagnosis, and a biopsy of intraabdominal adipose tissue, which showed only accumulation of macrophages. The patient had no symptoms at this time, and examination was therefore discontinued with a tentative diagnosis of post-thrombotic syndrome following deep vein thrombosis and essential thrombocythaemia.
Venous thromboembolism, which includes deep vein thrombosis and pulmonary embolism, is a common diagnosis both in general practice and in hospitals. The most common risk factors are hereditary thrombophilia, malignancy, immobilisation, trauma, pregnancy, use of oestrogens, previous venous thromboembolism and antiphospholipid syndrome (1, 2). There is also an association between thromboses, both arterial and venous, and essential thrombocythaemia (3), as in our patient. Post-thrombotic syndrome – characterised by pain, a sensation of heaviness, oedema, rubor and varicose veins – occurs in approximately 20–50 % of patients with venous thromboembolism and is the most common late complication of deep vein thrombosis (4). Our patient had several clinical features that were inconsistent with classic post-thrombotic syndrome. However, clinical findings can be non-specific and variable, making a definitive diagnosis challenging.
Mutations in the JAK2 gene are important in the pathogenesis of several myeloproliferative neoplasias. The mutation makes haematopoietic cells more sensitive to growth factors such as erythropoietin and thrombopoietin, leading to increased cell proliferation and potential malignancy. The mutation has been detected in almost all cases of polycythaemia vera, while it is present in about 50–60 % of patients with essential thrombocythaemia and primary myelofibrosis (5). Chronic myeloid leukaemia, the fourth of the myeloproliferative neoplasias, does not have the same association with the JAK2 mutation, but is characterised by the so-called Philadelphia chromosome (6).
One year later, the patient experienced recurrent and prolonged episodes of fever and dry cough. Upon suspicion of pneumonia, she was treated with repeated courses of various antibiotics, but with either no effect or only a short-term effect. She gradually became increasingly dyspnoeic and developed ascites, and was admitted to a local hospital for further examination. CT abdomen was performed again, along with MRI pelvis and CT thorax, and showed reactive changes as previously, but now also with pleural and pericardial thickening. Blood and urine cultures and a nasopharyngeal test yielded no notable findings. Bronchoscopy with bronchoalveolar lavage was negative for infectious agents, while macrophages were detected upon ascitic fluid cytology. There was no suspicion of autoimmune disease, and a gynaecologist found no evidence for gynaecological cancer.
The CT abdomen images were sent to a university hospital for further evaluation. The radiologist noted striking changes around the kidneys (Fig. 1) – suggestive of so-called hairy kidneys. The patient was transferred to the university hospital for further examination.
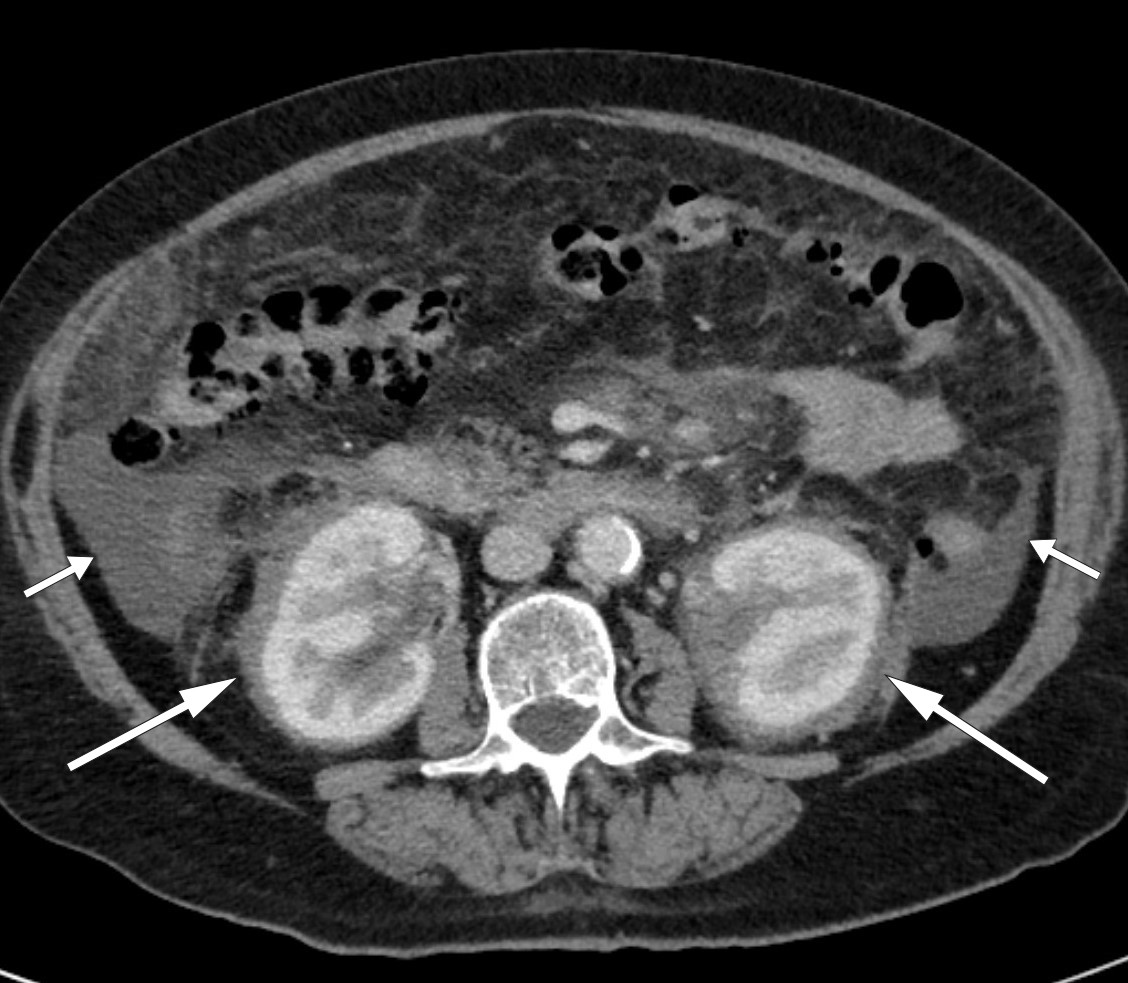
Figure 1 Ascites (short arrows) and symmetrical, oedema-like infiltration in perirenal tissue (long arrows) consistent with hairy kidneys on CT abdomen.
Thickening of the pleura and pericardium can be detected by radiological examination or echocardiography and may be seen in cases of, for example, infection, connective tissue disease, mesothelioma, lymphoma or metastasis (7, 8). If the pleura, pericardium and peritoneum are all affected, then systemic disease must be suspected.
‘Hairy kidneys’ describes retroperitoneal fibrosis that infiltrates perirenal adipose tissue, with thickening of the renal capsules. Retroperitoneal fibrosis is usually localised around the abdominal aorta and common iliac vessels, but may also be more widespread (9). More than 70 % of cases of retroperitoneal fibrosis are idiopathic (10), while the remainder are caused by factors including medications, malignancy, infections, surgery, radiation, amyloidosis and histiocytosis (9). Idiopathic retroperitoneal fibrosis is often classified as a chronic periaortitis, either isolated or in association with other autoimmune diseases or immunoglobulin G4 (IgG4)-related disease (11). Symptoms can be vague and non-specific. They typically include lower back pain, possibly radiating to the groin, accompanied by weight loss, reduced appetite, fatigue, fever, nausea and vomiting. It is not all that uncommon for the condition to be detected incidentally – as in our patient – during radiological examination of the abdomen. However, in our case, the infiltration was more widespread than is typical with retroperitoneal fibrosis.
The clinical picture was now dominated by fever, dry cough, dyspnoea and ascites. Blood samples taken upon admission to the university hospital showed elevated inflammatory markers with C-reactive protein (CRP) of 105 mg/l (reference range ≤ 5), sedimentation rate (SR) 98 mm/hr (1–28) and leukocytes 12.8 · 109/l (3.7–10.0). A leukocyte differential count showed 89 % (47–75) neutrophils, 7 % (21–43) lymphocytes, 3 % (4–14) monocytes, 1 % (1–4) eosinophils and 0 % (0–2) basophils. Haemoglobin was 9.7 g/100 ml (11.7–15.3), platelets 565 · 109/1 (145–390), creatinine 68 µmol/l (45–90) with estimated glomerular filtration rate (eGFR) 80 ml/min/1.73 m2, prothrombin time (PT)-INR 1.2 (0.8–1.2) and albumin 22 g/l (36–47). Potassium was slightly reduced at 3.1 mmol/l (3.5–4.4), whereas both sodium and calcium were within reference ranges with levels of 140 mmol/l (137–145) and 2.29 mmol/l (2.15–2.51) respectively. This was likely to be a systemic disease involving a number of organs and organ systems, and a comprehensive diagnostic examination was therefore initiated.
In addition to the perinephric changes, there were notable findings on CT thorax (Fig. 2) and full body scintigraphy (Fig. 3). A small pulmonary embolus was also discovered as an incidental finding. Cardiac MRI and echocardiography revealed locally thickened pericardium, while MRI head and orbita were normal. Abdominal paracentesis showed CD68-positive and CD1a-negative macrophages but no sign of microbes. In the ascitic fluid, which was translucent and yellow, the albumin level was 13 g/l and the protein concentration 24 g/l (below 30 g/l suggests transudate). The serum-ascites albumin gradient (SAAG) was 9 g/l, consistent with the ascites not being due to portal hypertension. The leukocyte count in the ascitic fluid was 0.20 · 109/l, which ruled out spontaneous bacterial peritonitis. A blood smear revealed granulocytosis and thrombocytosis, while a bone marrow smear showed reactive/normal bone marrow. A bone marrow biopsy revealed some weakly reactive megakaryocytes, while femoral biopsies showed only osteomyelosclerosis. A perinephric tissue biopsy showed fibrosis and histiocytes that were positive for CD68 and negative for CD1a, as well as a non-specific reaction for S100 proteins. Genetic testing for the BRAF mutation was negative.
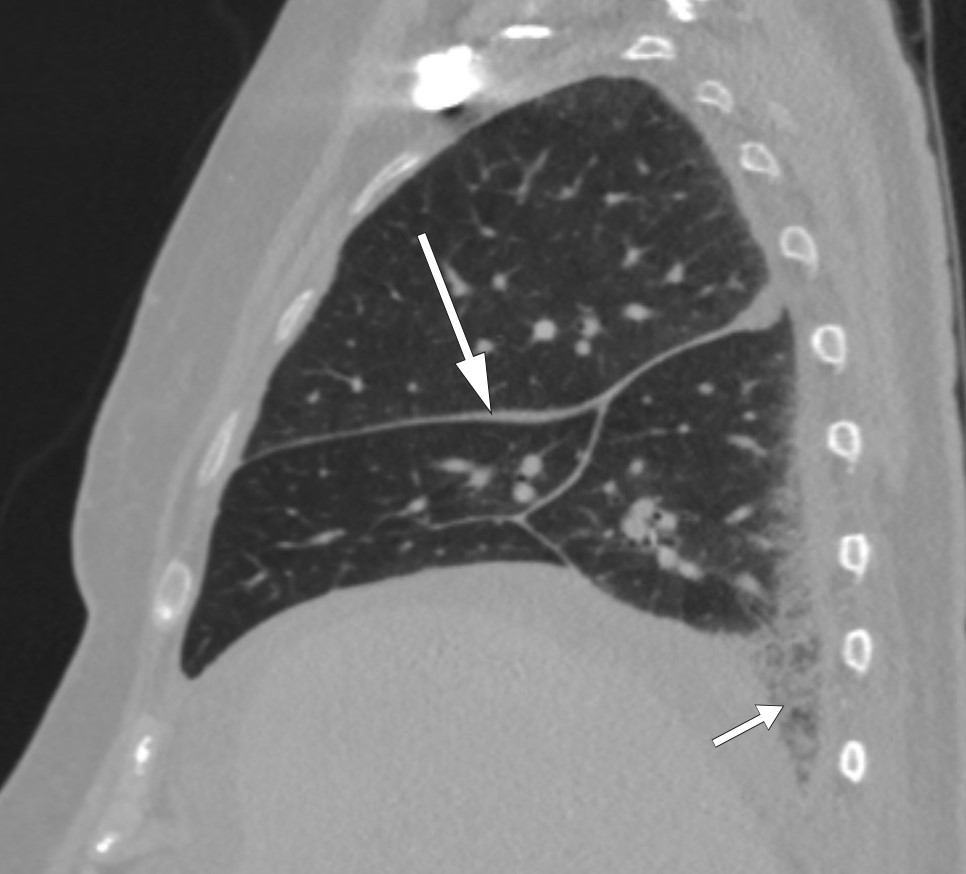
Figure 2 CT thorax showing even thickening of interlobular columns (long arrow) and reticular changes basally in the inferior lobe of the right lung (short arrow).
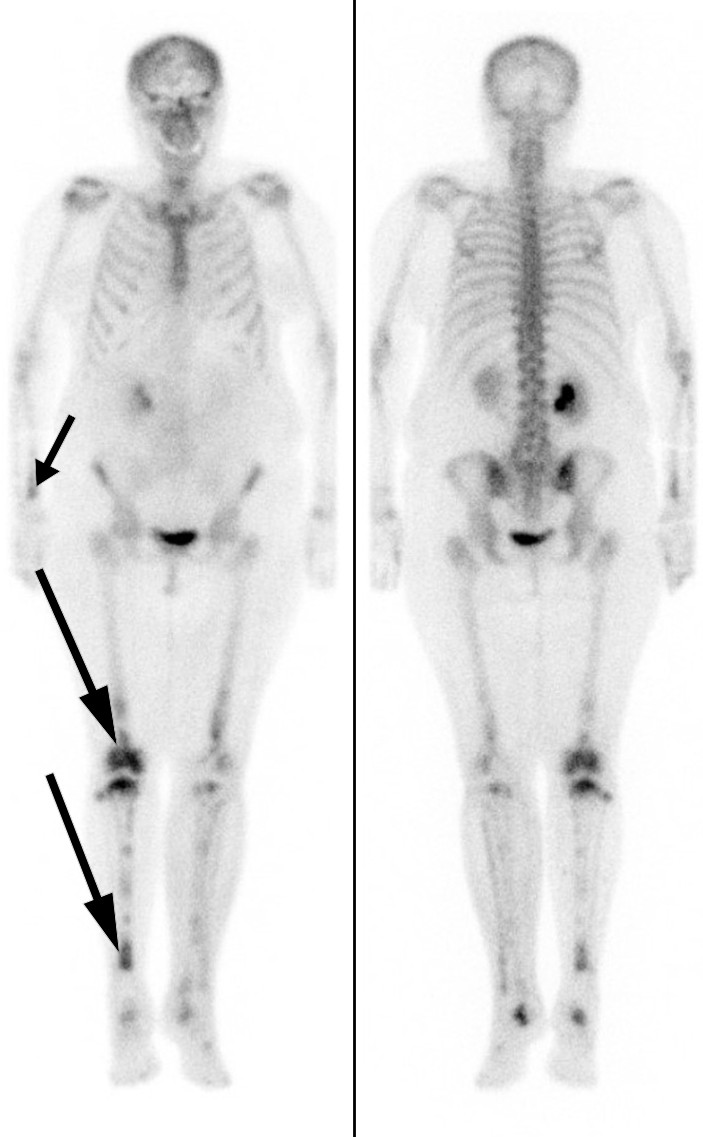
Figure 3 Skeletal scintigraphy with technetium-99 m showing increased radionuclide uptake in both lower extremities, especially in the right-sided diaphysis and metaphyses (long arrows). Note too the increased uptake in the right distal radius (short arrow).
In brief, the patient had general symptoms, respiratory symptoms and ascites. Inflammatory markers were elevated, and she had anaemia and hypoalbuminaemia. There were radiological findings perirenally, and in the lungs and skeleton, while macrophages and histiocytes were detected in the ascitic fluid and perirenal tissue respectively. She also had comorbid essential thrombocythaemia. The possibility that the patient’s current condition reflected a JAK2-V617F-positive myeloproliferative neoplasia was considered. Alternatively, she could have an unclassified myeloproliferative neoplasia (MPN), in which the essential thrombocythaemia had transformed into myelofibrosis, acute myeloid leukaemia or myelodysplastic syndrome, although this would not fully explain the condition. Widespread lymphoma and IgG4-related disease were also considered, as were medication side effects, but no evidence was found for any of these. Langerhans cell histiocytosis would be a potential explanation for the histiocytes. However, Langerhans cells express CD1a, whereas the patient’s histiocytes did not.
Immunohistochemistry and immunocytochemistry can be used to supplement the histological and cytological microscopic examination of tissue. The immunological method detects specific proteins inside the cell or in the cell membrane, and is important in the diagnosis of cancer, infectious disease and immunological disease. CD68 is expressed by monocytes and tissue macrophages (12), CD1a is often used as a marker of dendritic cells (13), while S100 comprises a group of proteins that are important in inflammatory diseases, cancer and the immune system (14).
Detection of mutations within cells can also be vital, both for diagnosis and treatment. Mutation of the BRAF gene, for example, leads to activation of an intracellular signalling cascade associated with a number of types of cancer, including malignant melanoma (15).
Several differential diagnoses were discussed. When the case was first discussed with the university hospital, the radiologist had pointed out the possibility of Erdheim-Chester disease (ECD), on the basis of the patient’s hairy kidneys. Clinical findings, blood tests, radiology and histology suggested that this was the most likely diagnosis. Treatment with interferon-α was initiated, but had no effect on disease activity, and the patient experienced side effects in the form of a flu-like illness. She was admitted to another university hospital, where combination therapy with prednisolone and the mTOR inhibitor sirolimus was initiated. Clinical improvement occurred after only a few days, and four months later she was significantly better. The fever and cough resolved, while sedimentation rate, leukocytes, haemoglobin and albumin were normalised. PET-CT showed regression of pathology. The patient is now being monitored with regular check-ups.
Discussion
Erdheim-Chester disease is a rare non-Langerhans cell histiocytosis characterised by infiltration of CD68-positive and CD1a-negative histiocytes in the long bones and retroperitoneum. Multiple organs may be affected, and the aetiology is unknown. Mutations in BRAF-V600E (16, 17), NRAS and PIK3CA (18), all of which are proto-oncogenes involved in the carcinogenesis of several types of cancer, may be important. This suggests that the disease may be a malignant disorder (19). There has also been discussion as to whether immune-mediated mechanisms, cytokines and chemokines may lead to accumulation of histiocytes in affected tissues, and whether this could indicate that there is an inflammatory component to the disease pathogenesis (20–22).
Histiocytoses can be divided into five groups, in which the common denominator is the accumulation of cells derived from dendritic cells or macrophages. More than 100 different types have been described, with a wide variety of symptoms, clinical features and histology (23). Erdheim-Chester disease belongs to the so-called L group, along with Langerhans cell histiocytosis. This group also features a combined form, as well as a form distinct from both Erdheim-Chester disease and Langerhans-cell histiocytosis on the basis of cell characteristics.
The diagnosis of Erdheim-Chester disease is based on clinical, radiological and histological findings. The most common manifestation involves the bones, central nervous system, kidneys and cardiovascular system (24, 25). Orbital infiltration has also been described, for example, in a case report in the Journal of the Norwegian Medical Association from 2014 (26). Symptoms depend on which organs are affected, but typically include skeletal or joint pain, dyspnoea, cough, exophthalmos or xanthomas as well as visual disturbances, ataxia, headache or other neurological symptoms. Cardiac involvement is often asymptomatic, but can have serious consequences. Pericardial disease such as pericarditis, pericardial fluid or tamponade affects up to 45 % of patients. Histiocyte infiltration of myocardial and coronary vessels occurs more rarely, but can result in ischaemia, valvular dysfunction or conduction abnormalities (27). Our patient had thickened pericardium – visualised on CT thorax, cardiac MRI and echocardiography – but no pericardial fluid and otherwise good cardiac function.
Symmetrical osteosclerosis in the diaphyses and metaphyses of the long bones, inflammatory changes around the aorta (coated aorta) and perirenal infiltration of histiocytes (hairy kidneys) are typical radiological findings (25, 28, 29). The latter is almost pathognomonic for the condition and was a key reason for the diagnosis being suspected early on in our patient. Histology is characterised by lipid-filled foamy histiocytes, often in combination with fibrosis. Immunohistochemically, these will be positive for CD68, CD163, and factor XIIIa, and in some cases for S100, which distinguishes them from histiocytes in a number of other conditions. They will also be negative for CD1a and Langerin (CD207) (27).
In addition to a number of typical features of the disease, our patient had ascites as a prominent symptom. This is rare in this condition, and was probably the result of hypoalbuminaemia secondary to prolonged inflammation and local intraabdominal inflammation.
Owing to the low incidence and prevalence of the disease, there is a lack of large randomised controlled trials of therapeutic options. Based on the studies that have been performed, the first-line treatment is interferon (IFN)-α-2a or pegylated interferon-α (PEG-IFN-α). Other alternatives are chemotherapy, anti-cytokine therapy, glucocorticoids or the BRAF-inhibitor vemurafenib. The latter is particularly appropriate when a BRAF mutation has been detected. Radiation and surgery may be suitable for palliative treatment (27).
Given that ascites was a key clinical finding, our case report describes an unusual presentation of Erdheim-Chester disease. Correctly diagnosing this disease requires first considering it as a possibility, especially when characteristic radiological features are present.
