About 150 persons develop amyotrophic lateral sclerosis (ALS) each year in Norway and this number is increasing (1). Traditionally, the condition has been regarded as a purely motor disorder characterised by progressive paralysis, but up to half of patients also develop signs of cognitive failure with behavioural and personality disturbances. A minority develop frontotemporal dementia (2). Patients typically die of respiratory failure within 2–3 years of diagnosis (3).
Amyotrophic lateral sclerosis is often divided into a sporadic form with no other known cases in the family (90–95 % of cases) and a familial form (5–10 % of cases). The latter usually shows autosomal dominant inheritance with incomplete and often unknown penetrance. Since the discovery of mutations in the SOD1 gene in 1993, pathogenic mutations have been detected in some 30 other genes in familial amyotrophic lateral sclerosis (3). These mutations account for about 60–80 % of familial cases in total (4). However, such mutations are also present in about 10 % of patients with sporadic amyotrophic lateral sclerosis and in an even greater proportion of those with concurrent frontotemporal dementia (5). The most common cause of both hereditary amyotrophic lateral sclerosis and frontotemporal dementia is an expansion of the GGGGCC hexanucleotide repeat in the C9orf72 gene (6). Testing for this mutation is not performed in Norway. Its frequency is therefore unknown, but in other countries it is reported in about 40 % of cases of familial amyotrophic lateral sclerosis, 10 % of sporadic amyotrophic lateral sclerosis and 20 % of frontotemporal dementia (7, 8).
There are currently no specific therapies for any of the genetic variants of amyotrophic lateral sclerosis. In the sporadic form, a positive test has uncertain significance for the disease risk among relatives and can prove a major additional burden for patient and relatives alike. In 2004, 2007 and 2012, the European Federation of Neurological Societies (EFNS) and the European Network for the Cure of Amyotrophic Lateral Sclerosis (ENCALS) therefore discouraged the genetic testing of patients with the sporadic variant, but were more open to the testing of persons with the familial type or with atypical clinical findings where testing could help clarify the diagnosis (9–11). In Norway, there are no official professional guidelines for the testing and follow-up of patients with amyotrophic lateral sclerosis. Recommendations in the ‘Neuro-Norwegian electronic medical handbook’ are consistent with those of EFNS. The purpose of this study was to investigate whether genetic testing practice for amyotrophic lateral sclerosis follows national and international recommendations.
Method
We searched the electronic health records at Akershus University Hospital for patients coded G12.2 according to ICD-10 (International Statistical Classification of Diseases and Related Health Problems) in the period 31 December 2004 to 31 December 2014. G12.2 encompasses motor neuron diseases, of which amyotrophic lateral sclerosis is by far the largest patient group. Congenital and hereditary variants of spinal muscular atrophy are excluded from G12.2. We evaluated whether the diagnosis was correct and whether the patient had primarily been examined and followed up at Akershus University Hospital. From the medical records we retrieved information on sex, age, family history, genetic testing and genetic counselling. The data were stored on secure research servers with de-identified clinical data stored separately from the key.
There is no clear consensus definition of what constitutes familial amyotrophic lateral sclerosis (12). In line with recent recommendations based on overlapping clinical findings and aetiology, patients with at least one first or second degree relative with amyotrophic lateral sclerosis or frontotemporal dementia were classified as having familial amyotrophic lateral sclerosis (13, 14).
The study was approved by the Data Protection Officer at Akershus University Hospital as a quality assurance study, removing the need to seek approval from the Regional Committee for Medical and Health Research Ethics (13, 14).
Results
The results of the medical record review are shown in Figure 1. Following exclusion of nine incorrectly coded patients, a total of 126 patients with amyotrophic lateral sclerosis were assessed at Akershus University Hospital in the study period. Of these, 115 were primarily examined and followed up at Akershus University Hospital, while 11 patients with the familial type and proven ALS-associated mutations were referred from other hospitals.
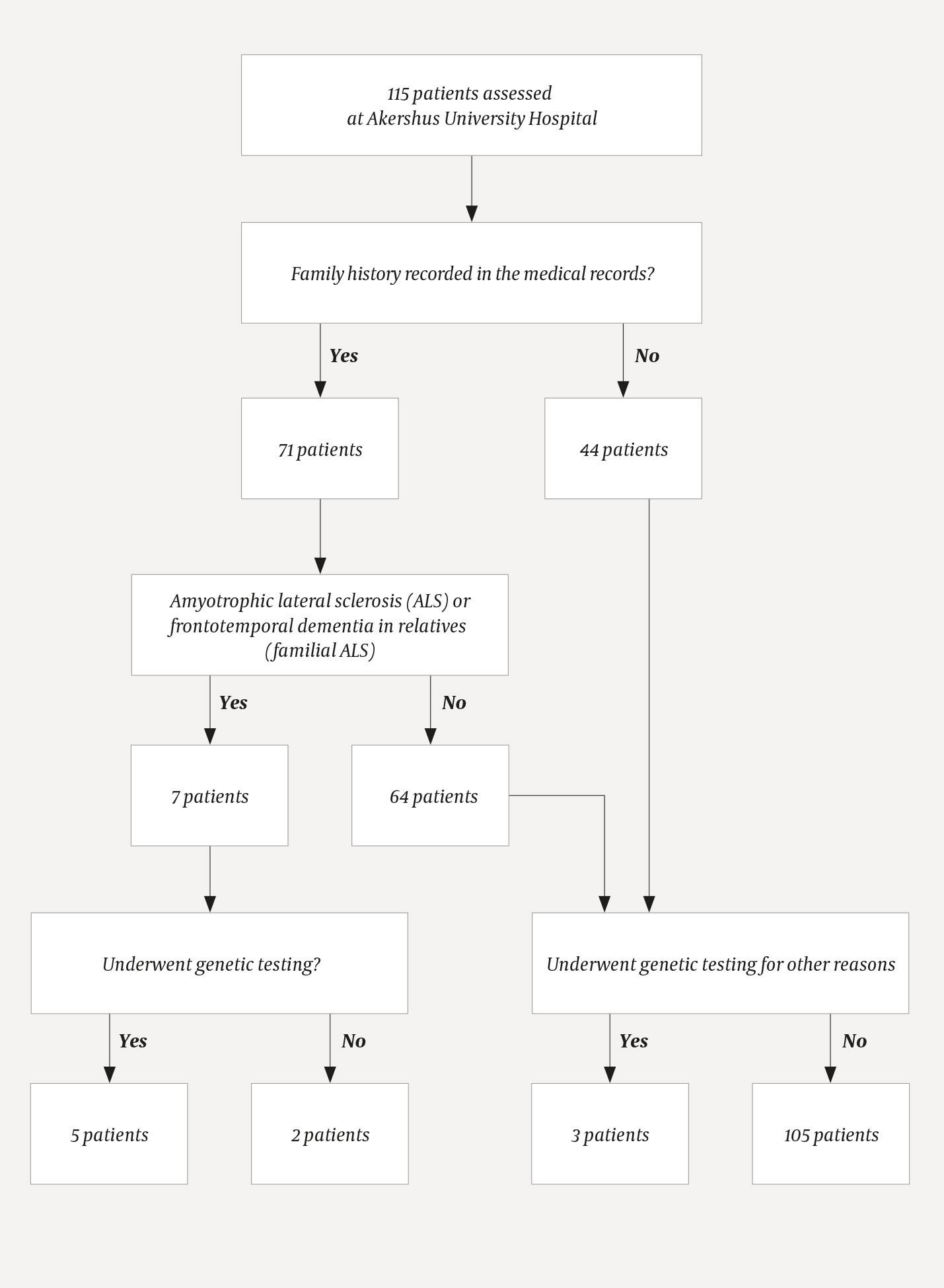
Figure 1 Genetic testing of patients with amyotrophic lateral sclerosis performed by Akershus University Hospital in the period 2004–14
Upon review of the medical records, we found that family history was recorded in 71 (62 %) of the 115 patients who had been assessed at Akershus University Hospital. Of these 71, seven (10 %) had close relatives with amyotrophic lateral sclerosis or frontotemporal dementia, consistent with familial amyotrophic lateral sclerosis. Of these, genetic analysis had been performed in five patients. No mention was made in the medical records as to whether genetic testing was considered for the other two patients with the familial variant.
A blood test was sent for diagnostic genetic testing in the case of three patients with sporadic amyotrophic lateral sclerosis. Testing of these patients was justified on the basis of an atypical disease course or another serious neurological disease (not motor neuron disease or frontotemporal dementia) in close relatives, or testing was performed at the request of a specialist in medical genetics in connection with the relatives’ wish for predictive testing.
Genetic testing was performed almost exclusively on the SOD1 gene, and mainly in the second half of the study period. With the exception of one patient in whom a heterozygous mutation of uncertain significance was found, all patients in whom an ALS-related mutation was detected were offered genetic counselling. In consultation with the patient, relatives were also informed and offered genetic counselling. Genetic counselling was not offered to any other patients, and nor was it mentioned in the medical records of the two patients with suspected familial amyotrophic lateral sclerosis who did not undergo genetic testing.
Towards the end of the study period, patients with a proven ALS mutation were invited to participate in research projects. Five donated skin biopsies for the generation of induced pluripotent stem cells. The patients were also given contact details that would open up the possibility of participating in a treatment study abroad.
Discussion
Our findings show consistently restrictive practice for genetic testing in cases of amyotrophic lateral sclerosis. Only three patients with the sporadic variant underwent diagnostic genetic testing, and justification for this was provided in all cases in the medical records. With the exception of one patient with the sporadic variant in whom testing was initiated by a specialist in medical genetics based on the desire of relatives for predictive testing, our practice was in line with European recommendations that specifically discourage genetic testing in typical cases of the sporadic form (9–11).
Two out of seven patients with the familial variant were not offered genetic testing or genetic counselling. Since the issue was not discussed in the medical records, we do not know the reasons why genetic testing was not performed. The absence of any explicit justification may reflect limited awareness about the importance of heredity, or uncertainty regarding access to genetic testing and counselling. This assumption is supported by a study from 20 countries showing that neurologists with extensive experience of amyotrophic lateral sclerosis offer genetic testing more frequently than their less experienced colleagues (14). Another explanation may be a conscious or unconscious desire not to contribute to increased concern that relatives will also develop the disease. The fact that family history was not recorded in the medical records of almost 40 % of patients is consistent with both of these explanations.
The limited use of genetic testing may also reflect the limited provision of such testing in Norway. A number of other ALS-related genes, including C9orf72, which is by far the most common genetic cause of amyotrophic lateral sclerosis, were discovered during the study period. However, Oslo University Hospital tested SOD1 alone throughout the study period, and indeed other ALS-related genes are still not tested today. Mutations in SOD1 account for only a small proportion of familial amyotrophic lateral sclerosis cases (approximately 20 %), and testing of this gene alone is seen as insufficient both in cases of the familial variant and in cases of the sporadic type with atypical disease course (14, 15). The genetic epidemiology has not been mapped in Norway, but by testing C9orf72, FUS and TARDBP in addition to SOD1, the genetic basis of about two-thirds of cases of familial amyotrophic lateral sclerosis and 10 % of cases of the sporadic type could probably be identified (14, 16).
The discovery of new ALS-related genes may lead patients, relatives and doctors to view genetic testing as more worthwhile, as it will mean that testing is more likely to be able to identify or rule out genetic causes of the disease (17). Telemark Hospital now analyses 17 other ALS-related genes besides SOD1, as well as a number of genes associated with other motor neuron diseases, but does not currently test for C9orf72 (Helle Høyer, personal communication).
The main argument for not offering genetic testing in cases of the sporadic variant is that doing so poses a threat to the patients’ right not to know, as well as consideration for the relatives’ wish to remain ignorant of their disease risk (15). The discovery of an ALS mutation in an ALS patient has far-reaching consequences for healthy relatives. They must deal with the fact that they may be at significantly increased risk of developing the disease and must decide whether they would like predictive testing, which under Norwegian law requires genetic counselling. The uncertainty over the significance of both positive and negative results makes it difficult for healthy relatives to decide whether to undergo testing, as testing will not provide them with an unambiguous answer regarding their own risk (18). This differs from Huntington’s disease, where a negative genetic test rules out the disease while a positive finding also provides important information about anticipated prognosis (19).
The relatively restrictive practice of reserving genetic testing for patients with the familial type or an atypical disease course has been challenged in recent years (20), in part because it deprives the patient of the possibility of identifying the cause of their disease (15). In addition, the frequent discovery of ALS mutations in patients with an apparently sporadic form of the disease renders the distinction between familial and sporadic forms more arbitrary than previously assumed (20, 21). The relatively high likelihood of detecting an ALS-related mutation is thus used as an argument for both restrictive and liberal genetic testing practices in sporadic amyotrophic lateral sclerosis (11, 17, 20). Increased insight into the genetic causes of disease is important for understanding disease mechanisms, and many future treatment studies will probably require molecular testing (22). In our experience, many patients wish to participate in clinical trials, and it is therefore likely that the number of patients who request genetic testing will increase. Given that it will be possible to test for a greater number of genes, the chances of detecting gene variants with uncertain disease association and thus uncertain significance in terms of disease risk in relatives will also increase. There will thus be a markedly greater need for genetic counselling.
Many patients wish to know the cause of their own disease and the risk of it being passed on in the family (18). In a survey of 5 591 patients with ALS in the USA, 83 % of respondents stated that genetic testing should be offered to all patients with the disease, with 73 % believing that the benefits outweigh the drawbacks (23). Those who had undergone genetic testing reported mostly positive experiences, irrespective of the type of disease (sporadic or familial) and whether they had received genetic counselling (24). The response rate in this survey was low (8 %), and its generalisability is therefore uncertain. The discrepancy between the positive feedback and the restrictive practice reflected in our findings and in the European recommendations nevertheless raises the question of whether neurologists are adopting a paternalistic and overprotective approach, which perhaps reflects their own discomfort in engaging with these issues.
Conclusion
Our study revealed a restrictive use of genetic testing in amyotrophic lateral sclerosis. With the exception of the fact that family history was not recorded in almost 40 % of patients, the use of genetic testing was in line with European recommendations. Genetic testing did not include newly discovered ALS-associated genes, despite the fact that doing so could have increased diagnostic accuracy (17). A genetic diagnosis may open up the possibility of participating in treatment trials or studies of disease mechanisms (25), also for patients with mutations in SOD1, which appear to have a relatively high incidence in Norway (26). Several patients with proven mutations have donated skin biopsies for use in generating induced pluripotent stem cells and in studies of disease mechanisms, or have been offered the opportunity to participate in treatment studies abroad. The repertoire of genetic tests should therefore be expanded in line with international standards.
