A young woman contacted her general practitioner because of a lump in her left inguinal area, heart palpitations and increasing shortness of breath. During workup for possible systemic rheumatic disease, it was concluded that the symptoms were caused by an unusual condition.
A formerly healthy woman in her late twenties consulted her general practitioner (GP) because of heart palpitations. She had also noticed a lump in her left inguinal area. She was referred to a cardiologist, and an echocardiogram, exercise ECG, long-term ECG and carotid ultrasound were carried out with normal results. During the following months she became short of breath and failed to keep up with people of her own age when walking uphill. She therefore contacted her GP one year after the first consultation. It emerged that the patient for 2–3 years had noticed that the fingers of both hands became white on exposure to cold. Spirometry showed reduced forced vital capacity (FVC) 2.1 l (52 % of expected value), decreased forced expiration volume during the first second (FEV1) 1.9 l (50 % of expected value), with an FEV1/FVC ratio of 0.9.
Enlarged lymph nodes are a common reason for contacting a GP. The condition is usually transient, and the lump in her inguinal area was not further examined. Dyspnoea may be due to both cardiac and pulmonary conditions. The cardiologist found no evidence of cardiac disease. Spirometry revealed severely reduced vital capacity, decreased expiration volume and an increased ratio between expiration volume and vital capacity. This indicated a restrictive pulmonary disorder. In addition, the patient had three-phase Raynaud’s phenomenon, where her fingers whitened, then became cyanotic and finally reddened. The condition has a prevalence of 3–5 % and is due to vasospasm of the digital capillaries. Raynaud’s phenomenon usually occurs without any concurrent disease, but may be associated with rheumatic connective tissue disease (1). The patient’s medical history and the findings indicated a systemic rheumatic disease with severe pulmonary affection. The patient was referred to a pulmonary specialist and rheumatologist.
Two weeks later she was assessed at the Department of Pulmonary Medicine. She was found on examination to have unobstructed respiration at rest. Spirometry confirmed decreased FVC 2.1 l (52 %), FEV1 2.0 l (54 %) and slightly increased FEV1/ FVC 0.9. Measurement of gas diffusion showed reduced carbon monoxide diffusion capacity (DLCO) 4.7 SI (46 %). Body plethysmography demonstrated severely reduced total lung capacity of 3.1 l (58 %). During a six-minute walk test, oxygen saturation fell from 98 % to 79 %. A lung CT revealed peripheral coarse reticular markings that aroused suspicion of non-specific interstitial pneumonia (NSIP) of the fibrotic type (Fig. 1). Echocardiography revealed normal heart valves and pressures and an ejection fraction of 55 %. A 24-hour ECG and 5-day ECG monitoring revealed no signs of arrhythmia. Blood tests showed haemoglobin 12.9 g/dl (11.7–15.3), sedimentation rate 26 mm (1–17). C-reactive protein, thrombocytes and leukocytes were within the respective reference ranges. Alanine amino transferase (ALT) was 54 U/l (10–45 U/l), alkaline phosphatase (ALP) 52 U/l (35–105) and lactate dehydrogenase (LD) 310 (105–205 U/l). She tested negative for rheumatoid factor (RF), anti-cyclic citrullinated peptide (anti-CCP) and anti-neutrophil cytoplasmic antibody (ANCA). Anti-nuclear antibodies (ANA) were weakly positive, but with negative subgroups.
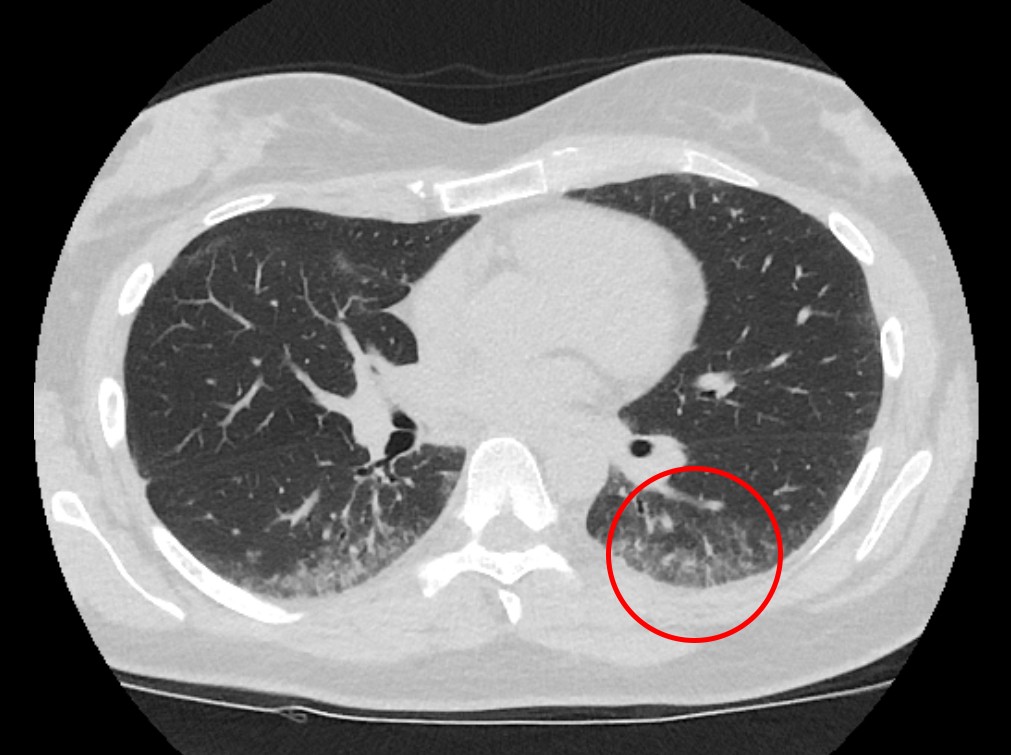
Figure 1 High-resolution thoracic CT shows peripheral subpleural micronodular lung opacities with some ground glass opacities.
Assessment of the patient’s dyspnoea showed pronounced restrictive ventilatory defect with reduced gas diffusion and significant desaturation during activity. The CT scan of the lungs indicated non-specific interstitial pneumonia. The main symptoms of this condition, which may be secondary to interstitial pulmonary disease, are dyspnoea and dry cough, and lung function tests find restrictive ventilatory defect and reduced gas diffusion (2, 3). The patient had a slightly elevated sedimentation rate which was assessed as non-specific. With the exception of elevated LD, her blood tests were normal. Rheumaserology showed a weak positive reaction to antinuclear antibodies, but more specific anti-bodies associated with rheumatic disease were not detected. The cardiac examination found no signs of arrhythmia, valve defects or pulmonary hypertension. Because the patient’s clinical findings of non-specific interstitial pneumonia, Raynaud’s phenomenon and positive anti-nuclear anti-bodies could be associated with a rheumatic condition, it was decided to await the results of the rheumatological workup.
A month later, the patient was examined by a rheumatologist, who found some tightness of the skin distally of the metacarpophalangeal joint, otherwise normal skin. Nailfold capillaroscopy revealed pathology, with several megacapillaries and some microhaemorrhages (Fig. 2). There were no signs of peripheral arthritis or muscular weakness. Blood tests showed increased creatine kinase (CK) 1 259 U/l (35–210) and normal metabolic test results. Urine test strips and microscopy were normal.
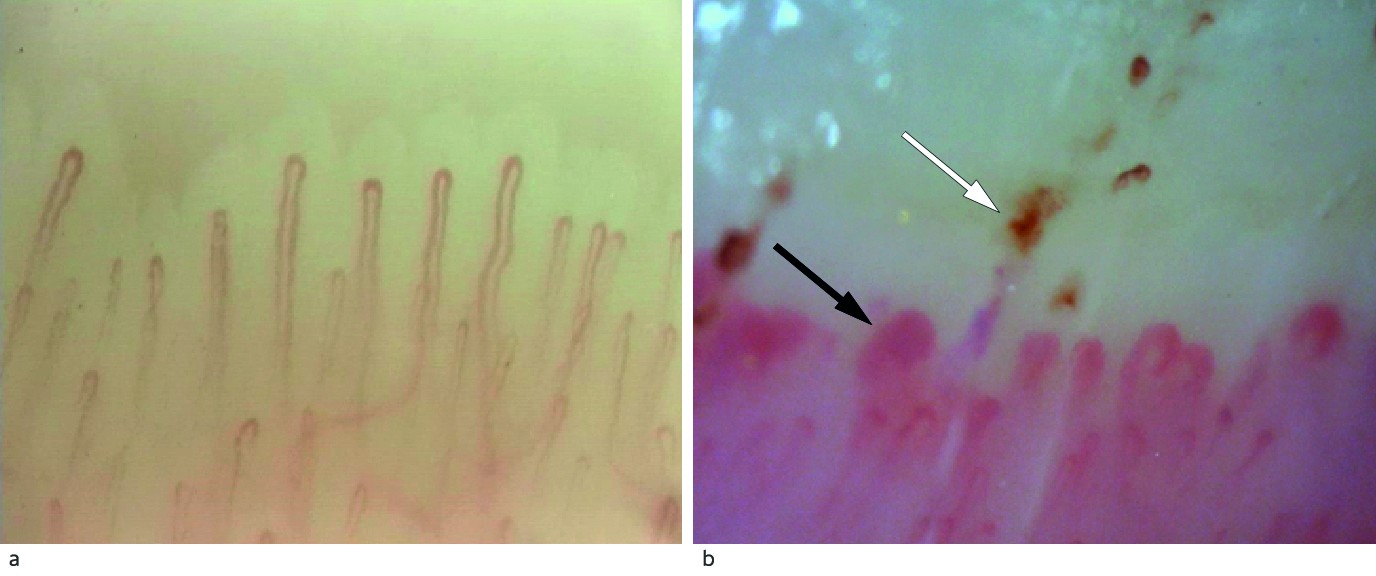
Figure 2 a) Nailfold capillaroscopy in a healthy person with normal capillary density and architecture. B) Capillaroscopy of our patient with considerable architectural derangement with megacapillaries (black arrow) and microhaemorrhages (white arrow). Photographer Øyvind Midtvedt, Rikshospitalet.
Triphasic Raynaud’s phenomena and oedematous, tight skin on fingers (puffy hands) are classic symptoms of systemic sclerosis (4). Nailfold capillaroscopy is used to distinguish between patients with primary Raynaud’s phenomenon and patients with interstitial pulmonary disease. Megacapillaries and microhaemorrhages indicated systemic sclerosis, but our patient’s fingers did not have oedematous, tight skin. Elevated CK is a non-specific finding, but myositis is usually associated with increased CK. The patient had no muscular weakness, which is a common finding in myositis. Both systemic sclerosis and myositis may be associated with interstitial pulmonary disease. The clinical picture was complex, but could indicate a scleroderma myositis overlap syndrome. The patient was referred to the regional department of rheumatology for a second opinion.
Three months after being assessed at the pulmonary department, the patient was hospitalised in the regional department of rheumatology for further diagnostics. Her general condition was good and the muscle strength of her extremities normal. There was no definite skin tightness, but significant pathological findings from nailfold capillaroscopy. A palpable lump in the left inguinal area was noted during clinical examination. Blood tests showed sedimentation rate 23 mm (1–17), elevated CK 810 U/l (35–210), troponin T 134 ng/l (< 14), LD 294 U/l (105–205) and negative rheumatoid factor and anticyclic citrullinated peptides. ANA was positive (> 160), with negative subgroups, including myositis- and scleroderma-specific antibodies and the malignancy-associated antibodies RNA polymerase III, TIF1-γ and NXP-2.
High resolution CT (HRCT) of the thorax showed unchanged subpleural micronodules with some ground glass opacities in both lungs, most pronounced in the lower lobes. Based on these findings sarcoidosis and hypersensitivity pneumonitis were suggested as possible differential diagnoses of non-specific interstitial pneumonia. On suspicion of myositis, magnetic resonance imaging (MRI) of the hip and thigh was carried out two days after hospitalisation. It revealed normal muscle tissue with no evidence of myositis, but with multiple enlarged lymph nodes in the left inguinal area and pelvis along the external iliac artery and vein.
Several findings made the diagnosis of scleroderma myositis overlap syndrome uncertain. The patient had no skin changes as in systemic sclerosis, but in rare cases systemic sclerosis can occur without skin involvement (5). The MRI scan did not reveal oedematous thigh musculature, which would be expected in a patient with active myositis. The absence of scleroderma myositis antibodies also reduced the probability of scleroderma myositis overlap syndrome. Sarcoidosis can cause muscular and myocardial affection. The clinical picture was atypical, with pulmonary changes, suspicion of myocardial affection and a palpable node in the left inguinal area. The patient was referred for a PET-CT and cardiac MRI for suspected cardiac involvement in interstitial pulmonary disease and a biopsy of the lymph node in her left inguinal area.
A week and a half after her discharge from the Department of Rheumatology, a PET-CT revealed slight to moderate uptake of 18F-fluorodeoxyglucose (FDG) in both lungs in known subpleural opacities, perceived as inflammation. Very high FDG uptake was found, consistent with multiple pathologically enlarged lymph nodes in the left inguinal area and left pelvis. PET-CT could not distinguish between severe inflammation and malignant activity (Fig. 3). An excision biopsy of a lymph node in the left inguinal area showed large cytoplasma-rich cells positive for melan-A-antigen and protein S100 consistent with metastasis from a malignant melanoma. A cardiac MRI demonstrated no signs of sarcoidosis or myositis-related myocarditis.
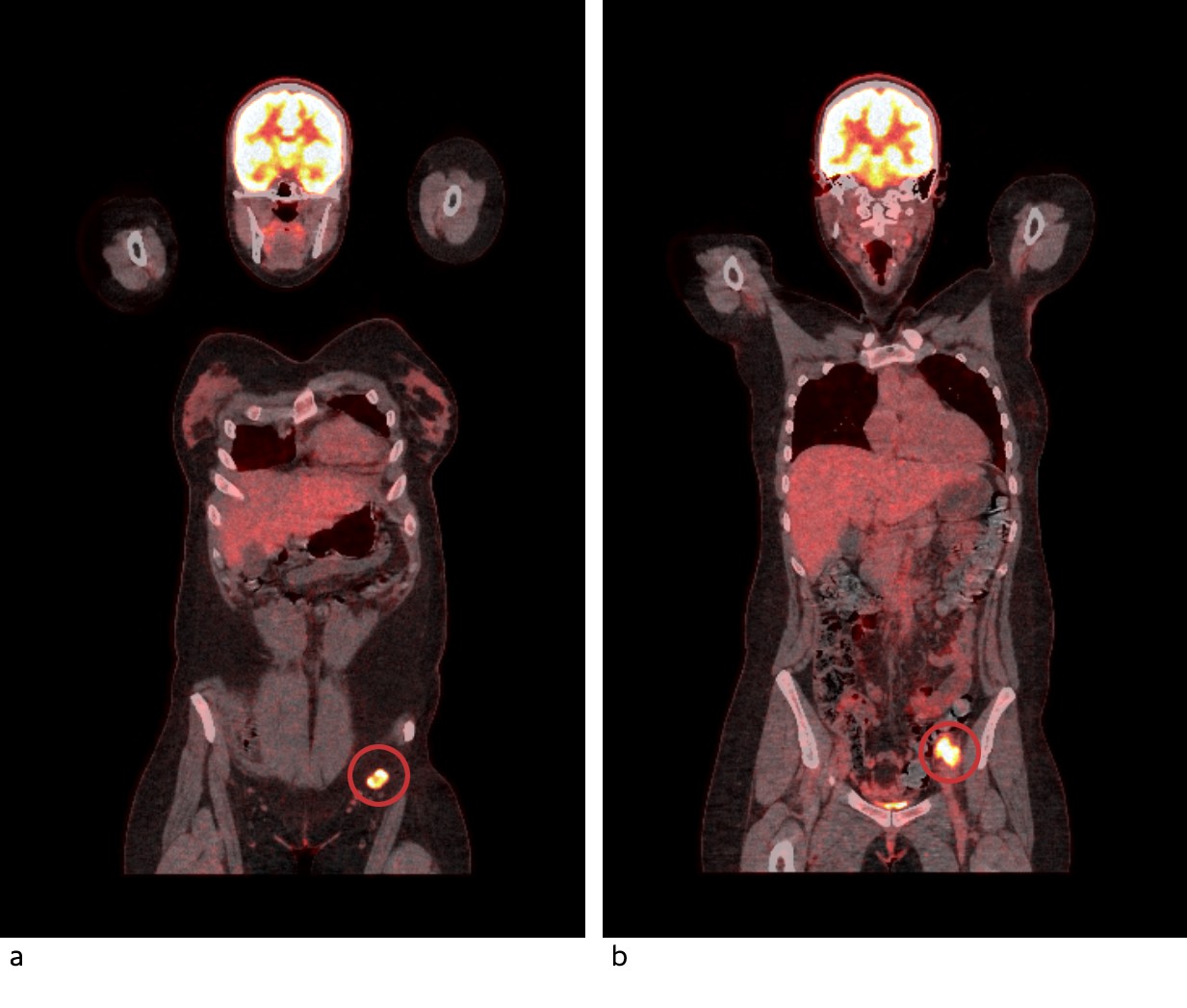
Figure 3 PET-CT shows high uptake of 18F-fluorodeoxyglucose in pathologically enlarged lymph nodes in the left inguinal area (a) and left pelvis (b).
The workup had detected metastasis from malignant melanoma in the left inguinal area. The patient had not previously had moles or other skin changes removed. She was referred to a dermatologist, ophthalmologist and gynaecologist for determination of primary tumour and to the Department of Plastic and Reconstructive Surgery for consideration of a lymph node dissection.
The dermatologist, ophthalmologist and gynaecologist performed workup without finding a primary tumour as the origin of the lymph node metastases. Ultrasound of the pelvis detected a 22 x 24 x 26 mm possibly metastatic lymph node conglomerate. An ultrasound-guided biopsy from the changes in the pelvis showed that there was also metastasis from a malignant melanoma in the pelvis. CT of thorax, abdomen and pelvis revealed pathological lymph nodes in the left inguinal area and pelvis from the inguinal ligament up to the aortic bifurcation. There was no evidence of metastases in parenchymatous organs or the skeleton.
The patient was discussed by a multidisciplinary team. She was considered to be in operable clinical stage III, with regional lymph node metastases without remote metastases. According to the current national guidelines for treatment of malignant melanoma, surgery is the first choice for operable stage III disease (6), and inguinal and pelvic lymph node dissection was recommended.
A week and a half after the last CT scan, and five months after she was referred to the specialist health services, the patient had surgery with combined open inguinal lymph node dissection and robot-assisted laparoscopic pelvic lymph node dissection. A histological examination of the specimen excised from the inguinal dissection showed a total of 16 lymph nodes and lymphoid foci with reactive changes. In the pelvic specimen, metastases from malignant melanoma were found in three of 36 lymph nodes. A molecular pathology examination of the excised specimen revealed mutation of the protein kinase BRAF.
The patient was perceived as being radically treated, with pathological stage IIIC. In line with current national guidelines, no indication was found for adjuvant systemic treatment for her melanoma. The rheumatic symptoms picture was perceived as a paraneoplastic condition.
At follow-up by the rheumatologist six weeks after the procedure, the patient reported improvement of her rheumatic symptoms. Because of the patient’s cancer, disease-modifying immunomodulatory therapy with rituximab or mycophenolate mofetil was not commenced, but prednisolone 30 mg x 1 by mouth in a tapering dose was recommended. Six months after the operation, the patient still had functional dyspnoea and Raynaud’s phenomenon, but felt generally better. Spirometry showed improved bellow function and gas diffusion with FVC 2.7 l (60 %), FEV1 2.3 l (61 %) and DLCO 5.5 (54 %). Recurrence of the cancer was not detected.
Discussion
Our patient initially had symptoms and findings consistent with a systemic rheumatic disease with pulmonary affection, and the workup aimed to clarify this. Initially, the palpable lump in the left inguinal area was not emphasised. At the time of the operation, the patient had felt the lump for about eighteen months. We must therefore assume that her cancer was present before she was referred to the specialist health services for other reasons. A targeted examination of the lump in her inguinal area at an earlier time would probably have led to the diagnosis malignant melanoma being made earlier.
Paraneoplastic syndromes are rare clinical syndromes that include non-metastatic systemic effects of malignancy that may cause neurological, endocrinal and rheumatic symptoms (7). The mechanisms have not been determined, but are assumed to be due to immune-mediated effects generated by tumour antigens (8). No specific tests can determine whether there is a paraneoplastic syndrome (7, 9). The concurrency of a rare malignant condition and symptoms of a systemic rheumatic disease that could not be classified as a specific condition led to our conclusion of a paraneoplastic syndrome. Myositis and systemic sclerosis are associated with malignancy. Patients with dermatomyositis and anti-TIFI-γ and/or anti-NXP-2 antibodies have a generally higher risk of cancer, and scleroderma patients with anti-RNA polymerase III have a higher risk of breast cancer (10, 11). Our patient tested negative for all these three antibodies, which strengthened our assumption that the rheumatic symptoms were an expression of a paraneoplastic syndrome and not a specific rheumatic condition (8, 9).
Treatment of underlying malignancy may lead to regression of paraneoplastic symptoms. Our patient had subjective and objective improvement of pulmonary function after the surgery for lymph node metastases. With interstitial pulmonary diseases, there will be components of both fibrosis and inflammation (3). Whereas the inflammation component is reversible, established fibrosis is non-reversible. We therefore do not expect the patient’s lung function to normalise.
An estimated 3 % of patients with metastasis from a malignant melanoma do not have a known primary tumour (12, 13). Metastases without a known primary tumour have been explained as immunologically conditioned regression of the primary tumour, that the primary tumour is still present, but so small that it cannot be detected, that the primary tumour has arisen from mucous membrane or that the melanoma has developed in a lymph node. Mutation analyses indicate that melanomas of unknown origin arise in skin, not mucous membranes (14). A meta-analysis found that patients with a metastatic melanoma without a known primary tumour had a better prognosis than patients with a known primary and the same tumour stage (15). The reason for this has not been determined, but the hypothesis of immunologically conditioned regression of a primary tumour may be consistent with the fact that this patient group has a stronger spontaneous anti-tumour immune response and therefore a better prognosis.
Early in the course of the disease, the patient was found to have increased lactate dehydrogenase. This is a non-specific test where values may be increased by myositis or acute cell damage in various organs. Values over the upper normal range are a negative prognostic factor for malignant melanoma, and form part of the staging of metastatic malignant melanoma (16). In patients with known metastatic melanoma, LD may be a marker of response to systemic treatment (17). In recent studies, adjuvant therapy with BRAF kinase inhibitors (18) and immunotherapy (19, 20) result in higher survival for stage III patients, but current national guidelines do not recommend adjuvant systemic therapy after radical macroscopic treatment of stage III melanomas (6).
First-line therapy for patients with stage IV or inoperable stage III melanoma is immunotherapy or ligand-blocking monoclonal antibodies against programmed cell-death protein 1 receptor (PD-1) (6). Immunotherapy may be associated with immune-related side effects with a risk of exacerbating an underlying systemic rheumatic disease (21). As with 40–50 % of patients with metastatic malignant melanoma, our patient had BRAF mutation. Systemic treatment with BRAF kinase inhibitors is not associated with immune-related side effects, and could therefore be an alternative to immunotherapy for our patient.
The case report illustrates the diagnostic challenges of a presentation of several apparently unrelated symptoms and findings. The case history is also a reminder that a malignant disease may give rise to symptoms from different organ systems.
