A man from a Mediterranean country had recurrent lymphadenopathy, fever and rash. This case report illustrates an unusual illness in Norway and the challenges associated with its diagnosis and treatment.
A man who was originally from a Mediterranean country but who had been living in Norway for many years, was referred by his general practitioner to a surgical outpatient clinic because of lymphadenopathy in the right axilla. This had been discovered by the patient about two weeks previously. He had no fever or other general symptoms. Examination revealed multiple enlarged lymph nodes, the largest of which was 3 cm in diameter. He also had pustules in the axilla and on his back, suggestive of a staphylococcal skin infection. Blood tests were normal with the exception of CRP, which was 15 mg/l (reference range <5 mg/l). Sedimentation rate was 11 mm/hour (<13 mm/hour). Axillary ultrasound revealed pathological lymphadenopathy. CT abdomen and thorax two weeks later showed multiple enlarged lymph nodes in the right axilla and several borderline size retroperitoneal lymph nodes, as well as a normal liver and spleen. Fine needle aspiration cytology of the largest lymph node three weeks after the first consultation revealed lymphoplasmacytoid cells and epithelioid cells. Ziehl-Neelsen cytological staining of fluid from the lymph node revealed no acid-fast bacilli, and a mycobacterial culture was negative. No antibodies were detected against HIV. The patient was treated with dicloxacillin tablets for the skin abscesses, which then disappeared. A lymph node biopsy was planned, but was later cancelled when the lymph nodes decreased markedly in size.
Skin lesions with small pustules may suggest skin abscesses with Staphylococcus aureus. The presence of infection can be confirmed using bacteriological culture. A bacterial infection such as this would result in lymphadenopathy. When enlarged lymph nodes cannot be attributed to an infection, cytology and histology of the lymph nodes are important to rule out lymphoma or cancer metastases. In persons from countries with a high incidence of tuberculosis, lymph node tuberculosis must also be ruled out with Ziehl-Neelsen staining and mycobacterial culture.
Two and a half years later, the patient was acutely hospitalised after two weeks of fever, chills, night sweats, nausea and vomiting. He had experienced involuntary weight loss of 10 kg over the past six months. Upon arrival, lymphadenopathy was again detected in the right axilla. Rectal temperature was 38.1 °C, but the results of a clinical examination were otherwise normal. Laboratory tests showed a sedimentation rate of 33 mm/h (<13 mm/hour), Hb 11.2 g/dl (13.4–17 g/dl), leukocytes 5.7 ∙ 109/l (3.5–11 ∙ 109/l), thrombocytes 219 ∙ 109/l (145–348 ∙ 109/l), creatinine 54 μmol/l (60–105 μmol/l), serum calcium 2.11 mmol/l (2.15–2.51 mmol/l), alkaline phosphatase 357 U/l (35–105 U/l), ALT 114 U/l (10–70 U/l), LDH 191 U/l (105–205 U/l), glucose 4.9 mmol/l (4–6 mmol/l), albumin 33 g/l (36–45 g/l) and CRP 121 mg/l (<5 mg/l). CT during admission showed lymphadenopathy in the right axilla, an enlarged spleen (13 × 15 cm), and an increase in the number and size of lymph nodes in the hilum of the liver, the retroperitoneum, along the pelvic blood vessels, and in both sides of the groin. The Mantoux test was normal, and serum ACE was 101 U/l (20–110 U/l). The test results gave no reason to suspect tuberculosis or sarcoidosis. A bone marrow biopsy from the iliac crest while in hospital provided no evidence of lymphoma or sarcoidosis, but did reveal some dysplastic features that were interpreted on the basis of clinical tests as reactive changes. A lymph node was removed from the right axilla, and the results of histological testing were described as follows: ‘Lymph node from the right axilla with peculiar vascular proliferation in the sinus and perilymphatic tissue.’ After 10 days, no definitive diagnosis had been made and the patient was discharged without treatment. Sarcoidosis was considered most likely even though levels of angiotensin-converting enzyme (ACE) were normal.
Sarcoidosis is often discovered as an incidental finding on chest x-ray. The symptoms are non-specific and include fatigue, dry cough, night sweats, fever and weight loss. The most frequent findings on chest x-ray are bilateral hilar lymphadenopathy and both diffuse and focal changes in the lung parenchyma. Quantification of ACE is the most important biochemical assay. Löfgren’s syndrome, which is an acute form of sarcoidosis, is characterised by bilateral hilar lymphadenopathy, erythema nodosum and joint pain. There are no definitive diagnostic tests for sarcoidosis (1). The tentative diagnosis of sarcoidosis in our patient was based on the clinical picture and exclusion of key differential diagnoses. Lymph node and bone marrow biopsies did not provide evidence of lymphoma. The lymph node biopsy was difficult to interpret, but was not sent for a second opinion.
Further outpatient testing with serum protein electrophoresis revealed no monoclonal components, which ruled out myelomatosis, and immunoglobulins within normal ranges. CT abdomen and thorax six months after the first hospitalisation showed progression of the lymphadenopathy in both axillae and in the abdomen. The patient did not wish to undergo another bone marrow biopsy, and therefore only sternal bone marrow aspiration was performed, which showed normal findings. Serological testing revealed a history of previous Epstein-Barr virus, cytomegalovirus and hepatitis B infections, but no evidence of syphilis, hepatitis A or C, toxoplasmosis or borreliosis. Mycoplasma pneumoniae-PCR and a repeat HIV test were negative, as were tests for rheumatoid factor (RF), antinuclear antibodies (ANA) and antibodies against cyclic citrullinated peptides (anti-CCP). A rheumatic disease such as rheumatoid arthritis was therefore unlikely.
Three years after the first consultation, the patient was acutely hospitalised with a cough. An x-ray suggested pneumonia, and treatment with penicillin G (5 million IU × 4 intravenously) proved successful. While in hospital, the patient developed renal failure with creatinine 471 μmol/l (60–105 μmol/l) and urea 38.2 mmol/l (3.5–8.1 mmol/l). No pre- or postrenal cause of the renal failure was found, and dialysis was not required. Renal biopsy revealed granulomatous interstitial nephritis, and corticosteroid therapy was initiated, first intravenous methylprednisolone (1 g daily for three days) and then oral prednisolone (60 mg daily with tapering). Steroid treatment led to recovery of renal function. Gastroscopy, which was performed due to a fall in Hb from 12.8 g/dl to 9.5 g/dl, was also normal. The patient was discharged with a tapering course of prednisolone.
Differential diagnoses included sarcoidosis, along with a potential drug-based or infectious origin of the lung disease and nephritis with renal failure. The patient’s medical history revealed that he had not started any new drugs recently, and renal biopsy failed to confirm an infection. It was concluded that the patient most likely had sarcoidosis-related nephritis.
The following year, the patient was followed up regularly at the outpatient oncology clinic. His prednisolone dose was reduced, but he was dependent on a maintenance dose of 10 mg daily to avoid symptoms of fever and lethargy. He gained 10 kg as a side effect of cortisone and developed hypertension that required medical treatment.
A year after starting high-dose prednisolone therapy, the man was acutely hospitalised again. This time he had a temperature of 40.2 °C, nausea and vomiting, and bilateral flank pain. He had very recently returned from a holiday in his native country. Antibiotic treatment was started with intravenous penicillin G and tobramycin. CRP increased to 260 mg/l and blood cultures showed growth of Salmonella enteritidis. Following resistance determination, the treatment was changed to ciprofloxacin (400 mg intravenously three times daily), and the patient quickly became symptom-free. One month later, he experienced a recurrence of his symptoms and was hospitalised again with salmonella septicaemia, which responded well to ciprofloxacin. A month after that, he was acutely hospitalised with joint pain, which was interpreted as reactive arthritis following a salmonella infection. He also had a somewhat unusual rash on his forearm, which was judged to be a possible side effect of prednisolone.
Two months after his last admission, the patient was hospitalised with symptoms of a respiratory tract infection without respiratory failure, and treatment with oseltamivir was started along with treatment for sepsis. Five days earlier he had been vaccinated against novel influenza strain (H1 N1). A nasopharyngeal culture was positive for H1 N1, indicating that he had not yet experienced an effect of the vaccine. He was discharged after a brief stay in hospital with instructions to continue oseltamivir at home.
After this episode, he never fully recovered. He was hospitalised yet again and underwent another gastroscopy, which revealed three reddish, pillow-shaped, haemangioma-like lesions in the stomach. The lesions were biopsied. When the macroscopic gastroscopy findings were compared with an endoscopy atlas, they were found to be pathognomonic for a gastrointestinal manifestation of Kaposi’s sarcoma (2). The pathologist was asked to examine the gastric tissue biopsies in light of this possibility. The patient also pointed out purplish skin lesions on his right forearm and left wrist that had increased in size over the last five months (Figure 1). The skin lesions on the arm were biopsied, and histological testing confirmed Kaposi’s sarcoma in these biopsies as well as those from the stomach (Figure 2). Both biopsies showed HHV8 (human herpesvirus 8)-positivity. The lymph node biopsy from five years earlier was re-evaluated and deemed also to be consistent with Kaposi’s sarcoma (Figure 3). Following the diagnosis, treatment was started with intravenous liposomal doxorubicin. Despite treatment, the disease progressed and spread to the liver, and the patient died just over two years after the diagnosis was made.
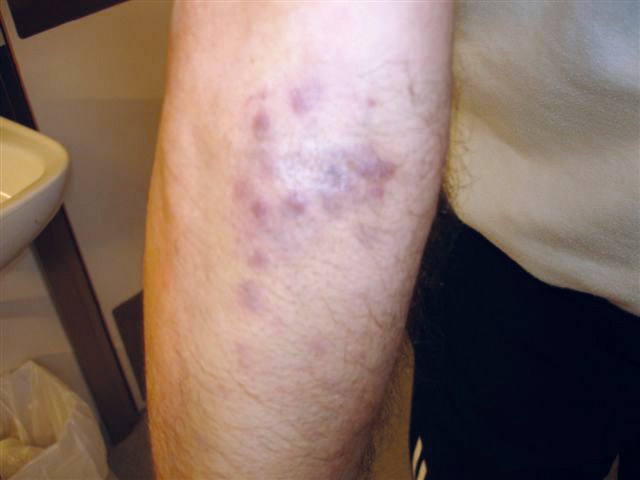
Figure 1 Photograph of the patient’s forearm showing purplish nodules in the skin.
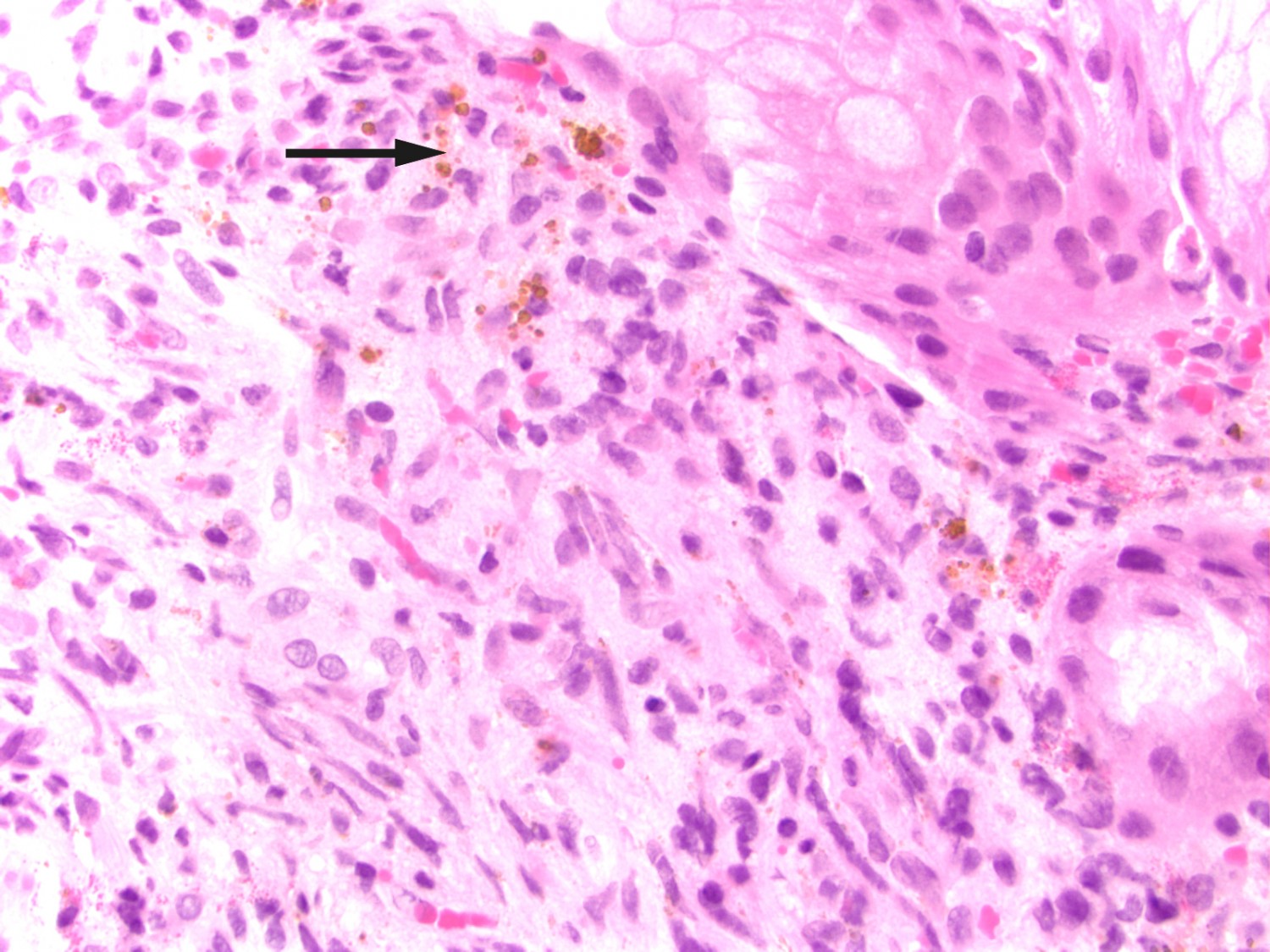
Figure 2 Biopsy from the gastric mucosa with capillary proliferation, extravasated erythrocytes and haemosiderin (arrow). Mitosis in a stromal spindle cell. HE-stained (haematoxylin-eosin).
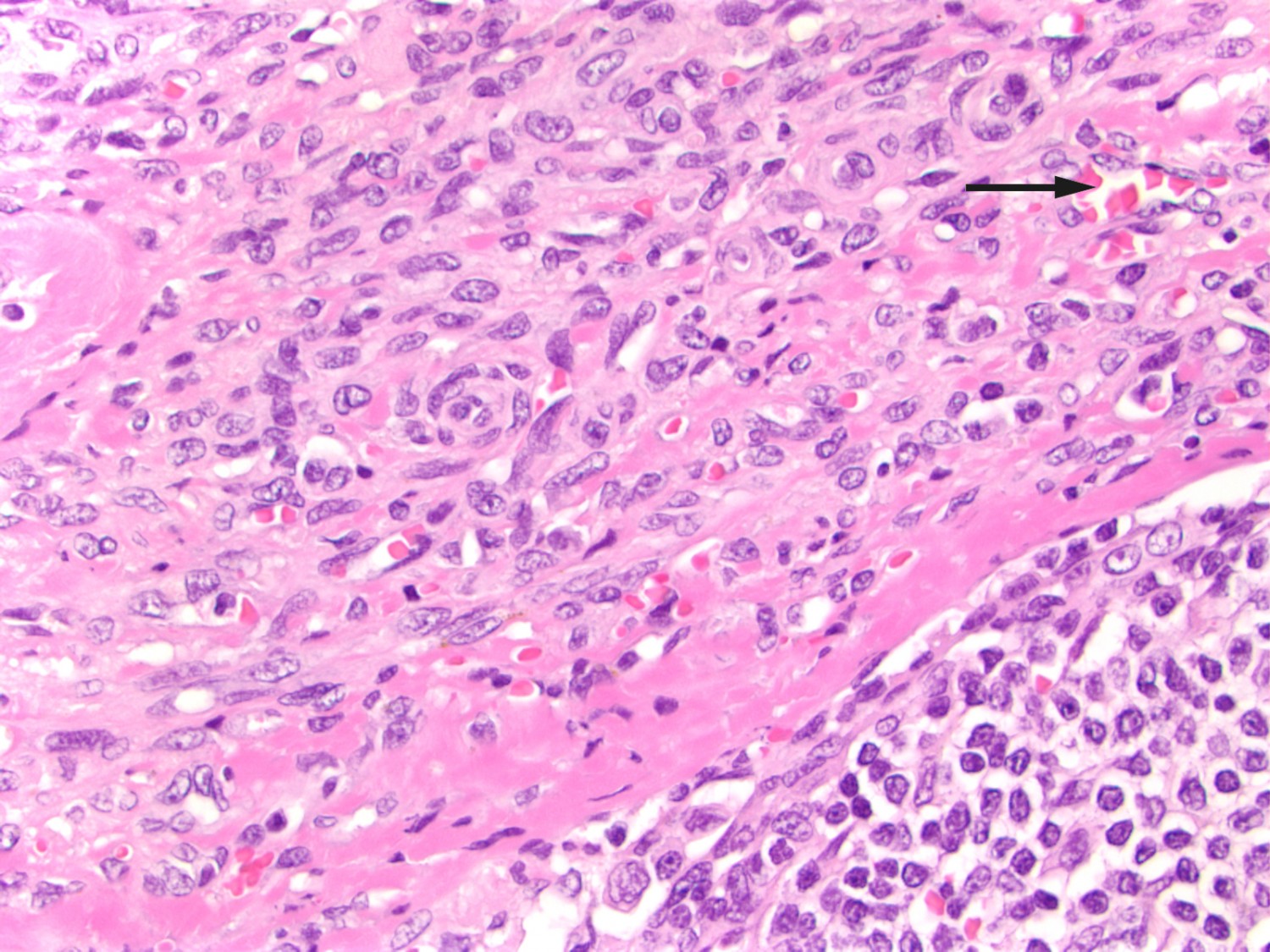
Figure 3 Lymph node with capillary proliferation. Spindle cells and extravasated erythrocytes in the capsule (arrow). Normal lymphoid follicle can be seen lower right. HE stained (haematoxylin-eosin).
Discussion
In our patient, recurrent fever and lymphadenopathy were the predominant symptoms and findings over a prolonged period. Sarcoidosis with renal involvement was suspected initially, as it was consistent with the patient’s medical history and no other likely diagnoses had been identified. After lengthy investigation, the patient was diagnosed with classic Kaposi’s sarcoma with involvement of the skin and viscera (stomach). Immunosuppressive therapy can be a contributing factor in the development of Kaposi’s sarcoma, but our patient first received treatment with cortisone after his disease picture was already established. Cortisone may nevertheless have contributed to the development of lesions of the skin and viscera (3). The patient was hospitalised with infections on numerous occasions over several years, but no signs of immune failure were ever detected. The renal biopsy was re-evaluated by a pathologist following the diagnosis of Kaposi’s sarcoma, but the pathologist was unable to find typical histopathological signs consistent with this disease. It is not certain that Kaposi’s sarcoma can account for the entire disease course in this patient, which also featured several serious infections and a diagnosis of interstitial nephritis of unknown origin. Some of these comorbid conditions are unlikely to be attributable to Kaposi’s sarcoma, but nevertheless create a confusing picture that complicates diagnosis. Five years prior to the diagnosis, a lymph node biopsy from the axilla had been described as ‘peculiar’, but no clear conclusion had been reached. Upon re-evaluation, the biopsy turned out to be consistent with Kaposi’s sarcoma. Perhaps the diagnosis could have been made earlier if this biopsy had been sent for a second opinion.
Kaposi’s sarcoma is a multifocal, low-grade, vascular tumour that may involve the skin, mucosa and viscera (3). The disease was first described by the Austrian doctor Moritz Kaposi in 1872 in five case reports, in which he referred to the condition as ‘idiopathic multiple pigmented sarcoma’ (4, 5). In the case reports he described oedema and purplish nodules on the skin, mostly on the hands and feet. He also performed microscopy of skin biopsies, and described small round cells (spindle cells), small haemorrhagic areas with nodules, and pigmentation (haemosiderin). All five patients died within 2–3 years.
In 1994, herpesvirus-like DNA sequences were identified in patients with AIDS-associated Kaposi’s sarcoma (6), and were named human herpesvirus 8 (HHV-8). HHV-8 is a member of the gamma herpesvirus family. Gamma herpesvirus causes tumours, lymphoproliferative disorders and lymphomas in humans and animals. HHV-8 is suspected to have spread to humans in Mediterranean countries from animals in Africa. Most primary HHV-8 infections are asymptomatic (3). The exact mode of virus transmission is unknown, but the virus can spread through sexual intercourse and is also found in saliva. Mother-to-child transmission of the virus has been demonstrated in Africa (7). HHV-8 is necessary but not sufficient for the development of Kaposi’s sarcoma (3).
Disease development is dependent on contributing factors such as chronic inflammation and immunosuppression (8). HIV indirectly promotes HHV-8 viral replication by suppressing the immune system and through the production of cytokines. Kaposi’s sarcoma is the most common tumour in patients with AIDS, but HIV alone cannot cause Kaposi’s sarcoma (6).
The prevalence of HHV-8 varies greatly, from high endemic areas in sub-Saharan Africa (30–70 %) via intermediate prevalence in Mediterranean countries (5–20 %) to low prevalence in Northern Europe and Japan (<5 %) (3, 9). HHV-8 prevalence reflects the incidence of Kaposi’s sarcoma in these countries. Only between three and nine cases of Kaposi’s sarcoma have been reported annually to the Cancer Registry of Norway (2003–2012) (10).
Kaposi’s sarcoma is divided into four types on the basis of epidemiology, underlying aetiology and clinical extent. There is extensive overlap between the different types, both in clinical symptoms and prognosis, but the more indolent cases feature mostly skin lesions, whereas the more aggressive forms more often involve the mucosa and viscera. Classic Kaposi’s sarcoma occurs most frequently in older men of Eastern European or Mediterranean origin. Endemic Kaposi’s sarcoma was the commonest form in Africa prior to the AIDS epidemic. In Uganda, Kaposi’s sarcoma accounted for 3–9 % of all cases of cancer in 1971. AIDS-associated Kaposi’s sarcoma has been the dominant form in Africa since the 1980s. AIDS-associated Kaposi’s sarcoma has a more aggressive course than endemic Kaposi’s sarcoma, but can be slowed or made to regress by effective HIV treatment. Iatrogenic Kaposi’s sarcoma is seen in immunosuppressed patients who have received organ transplants, especially in ethnic groups vulnerable to classic Kaposi’s sarcoma in Mediterranean countries (3). Our patient best fits into the category of classic Kaposi’s sarcoma, mainly on the basis of his origins and the absence of HIV infection or iatrogenic immunosuppression.
Histopathological findings are identical in the four types, with the most typical histopathological finding being spindle cells. The disease progresses in three histological stages: the first is the ‘patch stage’ and is characterised by flat, macular lesions. The next stage is characterised by plaques, while the final, tumour stage, is characterised by nodular lesions. These histological changes can easily be overlooked (5).
Treatment of Kaposi’s sarcoma varies depending on the clinical type and symptoms. In the absence of symptoms, treatment may be postponed. Relevant treatments are surgical excision, topical interferon alfa-2b, radiotherapy and systemic chemotherapy (5). Since there is no treatment that can eradicate HHV-8, it is debatable whether Kaposi’s sarcoma can be cured (11). Kaposi’s sarcoma limited to the skin normally responds well to chemotherapy, whereas nodular lesions are associated with shorter progression-free survival (12). There are few guidelines for treatment, and the disease course after various forms of treatment has mainly been described in retrospective, observational studies with diverse findings. After a median follow-up of 28 months, one study found that 55.5 % of patients showed disease progression, and 2.3 % had died from Kaposi’s sarcoma (12). In another study, median progression-free survival was 11.7 months following systemic chemotherapy, while half of all patients had died by 28.5 months (13). An Italian study found that only 12.2 % of patients with Kaposi’s sarcoma died from the condition. However, this was in a cohort of elderly patients (median age at death was 82 years for women and 85 years for men), and most died from cardiovascular disease (14).
Conclusion
This case study describes a protracted and challenging diagnostic work-up that eventually resulted in the patient being diagnosed with Kaposi’s sarcoma. Kaposi’s sarcoma is a rare form of cancer in Norway. Most cases occur in middle-aged men of Mediterranean origin and in patients with AIDS (10). Because the condition is rare and because both clinical and histopathological findings are non-specific, diagnosis can be challenging.
