A man in his seventies presented with complex symptoms and disease manifestations involving multiple organ systems. The symptoms would prove to have a common origin in a rare disease.
A previously healthy man in his seventies was admitted to Acute Admissions with elevated CRP levels, which had increased from 41 mg/l to 90 mg/l (<5) over three days, and an increased sedimentation rate of 51 mm/h (<20). Clinical examination revealed generalised macular eczema, which had first appeared two months earlier. He had had back pain 2–3 weeks prior to admission, but this was now slightly improved.
Two years before the current admission, the patient had been treated for L4/L5 lumbar spondylodiscitis, and had received appropriate antibiotic treatment for six weeks. He also had a history of repeated abdominal surgery following an inguinal hernia repair 14 years earlier. His most recent surgery was in the year prior to the current admission, for midline hernia and subileus. He was taking losartan for hypertension as well as folic acid supplements, but no other regular medication.
Given the previous medical history, new-onset back pain and elevated infection markers, the admitting doctor was concerned that the patient might have developed spondylodiscitis again.
Supplementary blood tests in Acute Admissions showed spontaneous prolongation of the prothrombin time with INR of 1.4 (<1.1) and a fall in CRP from 90 mg/l to 44 mg/l. Urine dipsticks showed positive results for leukocytes (2+), protein (2+), and nitrite.
Spondylodiscitis is a rare condition (1), for which the patient had received appropriate treatment two years previously. He was afebrile, and neither his medical history nor the clinical examination provided definite evidence of infection. He had no symptoms of peripheral neuropathy and no known history of trauma. His pain had been relieved by paracetamol, and recurrence of spondylodiscitis was considered unlikely. He had signs of coagulopathy, with spontaneously elevated INR, but his albumin levels were normal, which was inconsistent with a failure of hepatic protein synthesis (2). The patient had recently begun treatment for a biochemically confirmed folic acid deficiency, and it was thus possible that vitamin K deficiency attributable to malabsorption could have contributed to the coagulopathy. His rash was asymptomatic and unchanged since onset.
The patient was discharged from Acute Admissions with a diagnosis of urinary tract infection, which was treated with pivmecillinam 400 mg x 3 for five days. He was advised to contact his general practitioner for further follow-up. Two days after discharge, his urine culture was shown to be negative for bacterial growth.
Urinary tract infections are rare in men without underlying risk factors such as a urinary catheter, structural abnormalities of the urinary tract or neurological disease (3). Overall, there was no strong evidence for a urinary tract infection in this patient, and antibiotics should probably not have been given.
Two weeks after the patient had returned home from hospital, blood samples taken by his general practitioner showed new-onset anaemia with haemoglobin 11.5 g/dl (13.4–17). An automated differential blood count at the general practice generated a ‘blast alert’, and an additional blood smear was therefore performed, which was examined by a haematologist at the local hospital. The blood smear showed a slight left shift with some immature granulocyte precursors, but no sign of blasts.
A left shift refers to an increase in the proportion of immature granulocytes in the blood. A left shift may be reactive, as in cases of infection, but may also suggest haematological malignancy or bone marrow infiltration with an alternative aetiology (4).
Owing to unclear findings in the blood tests, the patient was referred to the haematology outpatient clinic, with his first consultation taking place one month after the assessment in Acute Admissions. By then his back pain had disappeared, but he described reduced general condition with fatigue and lethargy as well as weight loss of 3–4 kg over the past couple of months. He also reported symptoms including flatulence and loose stools after meals. Clinical examination revealed normal findings beyond the known asymptomatic rash on the torso and proximal extremities, which was a combination of brownish macular and erythematous exanthem. Supplementary blood tests showed haemoglobin 10.5 g/dl and a sedimentation rate of 64 mm/h. Liver tests were also elevated, with gamma-glutamyl transferase (γ-GT) 140 U/l (15–115), alkaline phosphatase (ALP) 278 U/l (35–105), activated partial thromboplastin time (APTT) of 57 s (30–44), and coagulopathy with INR of 1.4. Serum protein electrophoresis revealed an M-component, quantified as 2.7 g/l and classified as immunoglobulin M with associated free kappa light chains.
The patient had symptoms from multiple organ systems, particularly the skin and gastrointestinal tract, as well as notable weight loss and reduced general condition. He had a combination of abnormal liver test results, coagulopathy and anaemia. Rash and gastrointestinal symptoms could indicate coeliac disease. Carcinoid syndrome and vasculitis can also give rise to a similar clinical picture, with episodic gastrointestinal symptoms, increased sedimentation rate, and general malaise.
On account of his high sedimentation rate, anaemia, weight loss and altered general condition, the patient was referred for CT of the neck, thorax, abdomen and pelvis with intravenous contrast. This revealed small amounts of free fluid in the pelvis, splenomegaly of 17 cm (longest diameter), periportal oedema and prominent hepatic hilar lymph nodes, the largest with a short-axis diameter of 11 mm (Figure 1).
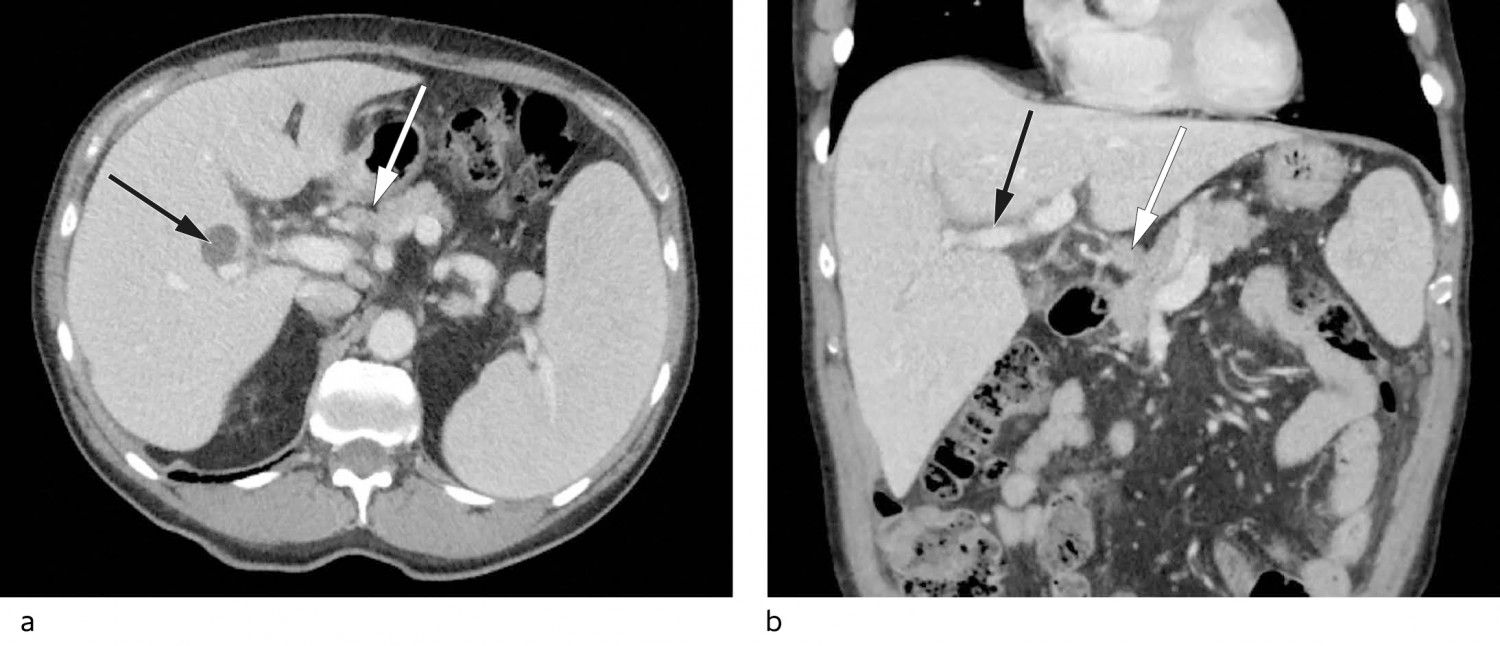
Figure 1 a) Axial CT of the upper abdomen with enlarged spleen and accessory spleen, prominent hepatic hilar lymph glands (white arrow) and a simple liver cyst in the right lobe (black arrow). b) Coronal CT with periportal oedema (black arrow) and prominent hepatic hilar lymph gland (white arrow).
Periportal oedema, the accumulation of fluid in tissues surrounding the portal venous system, is a nonspecific finding that may indicate liver pathology, but that can also be seen in extrahepatic diseases (5). A number of disorders can give rise to splenomegaly, including portal hypertension secondary to liver disease, as well as lymphoma and other haematological malignancies. However, lymphoma was less likely given the isolated finding of slightly prominent hepatic hilar lymph nodes in the absence of enlarged lymph nodes elsewhere. The anaemia, M-component, and left shift suggested haematological disease.
A few days after the initial consultation at the haematology outpatient clinic, bone marrow diagnostic tests were performed. A bone marrow smear showed an extremely cell-rich bone marrow, with a normal proportion of megakaryocytes and plasma cells, slight irregularities in erythropoiesis, and markedly left-shifted and increased granulocytopoiesis. The findings suggested a myeloproliferative disease. Crista biopsy showed a maximally cell-rich bone marrow with multiple infiltrating mast cells, some atypical (Figure 2). The bone marrow showed general hypercellularity, with predominant myelopoiesis and megakaryocytes with somewhat abnormal morphology. Immunohistochemistry showed that the mast cells were positive for mast cell tryptase, CD117 and CD25, while genetic analysis revealed a positive activating mutation in the KIT gene (D816 V). Levels of tryptase, a mast cell mediator, were also found to be elevated in serum, 116 mcg/l (< 20).
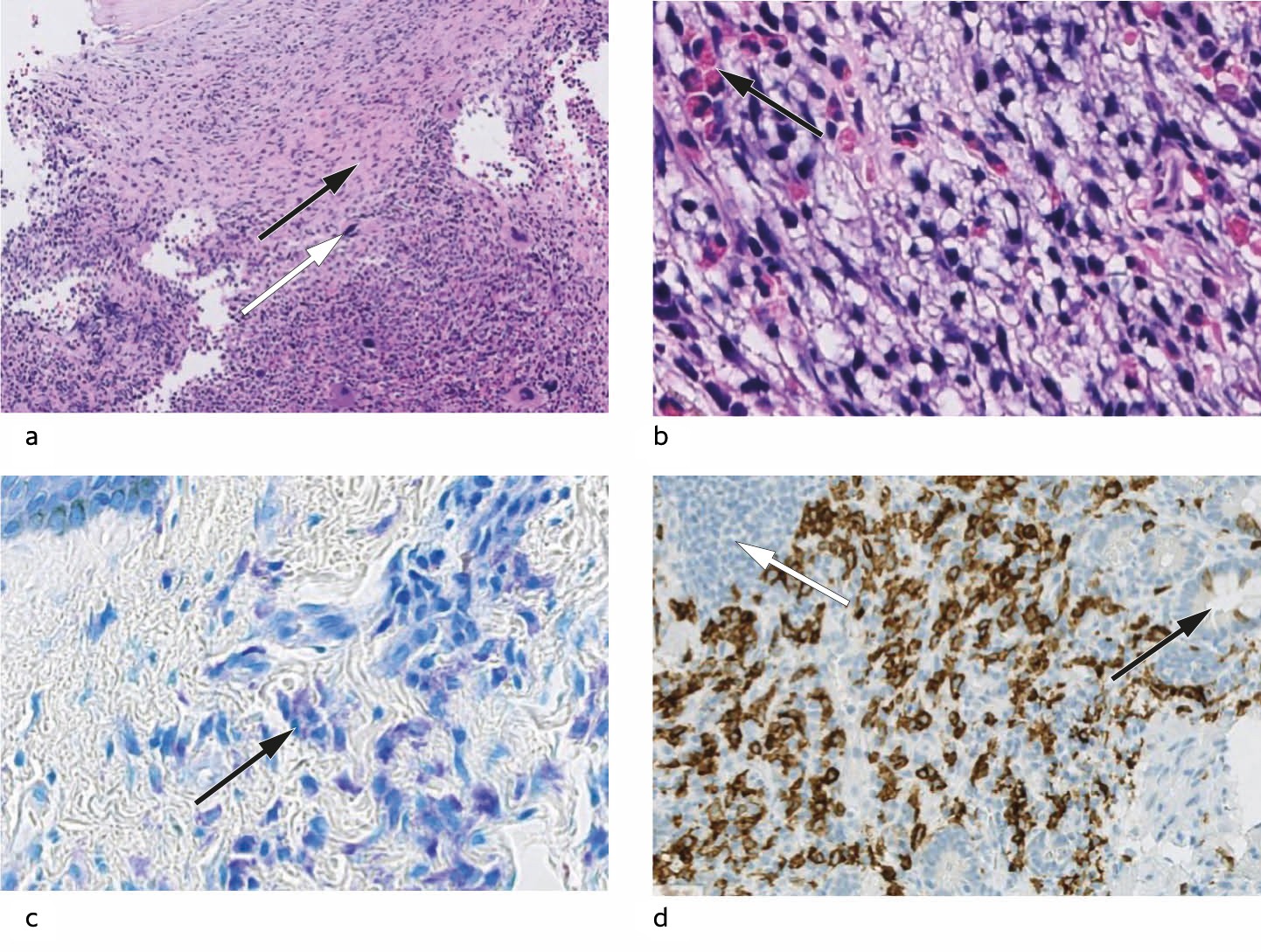
Figure 2 a) Bone marrow biopsy. The marrow is maximally cell rich. In the upper half, infiltrating mast cells can be seen in association with fibrosis (black arrow). In the lower half, hyperplastic haematopoiesis can be seen, with abnormal megakaryocytes (white arrow) (HE 150x). b) Bone marrow biopsy. Neoplastic mast cells with pale cytoplasm and hyperchromatic and irregular nuclei, interspersed with eosinophilic granulocytes (black arrow) (HE 400x). c) Skin biopsy. Giemsa-stained sections show accumulations of mast cells with typical basophilic granules in the cytoplasm of the upper dermis (black arrow). The basal layer of the epidermis can be seen in the top left corner (400x). d) Duodenal biopsy. Mast cell infiltration could not be seen clearly with routine staining, but immunohistochemical staining for CD117 resulted in brown colouration of the membranes of numerous mast cells in the basal layer of the mucosa. Crypts can be seen to the right (black arrow), and a normal lymphoid follicle can be seen top left (white arrow).
The bone marrow biopsy confirmed the diagnosis of systemic mastocytosis with an associated haematological neoplasm. Systemic mastocytosis can result in mast cell infiltration of multiple organs, and the patient therefore underwent further examination on account of his skin rash and gastrointestinal symptoms.
Two months later, a dermatologist performed a punch biopsy of the rash, which revealed mast cell infiltration of the dermis. A gastroscopy was performed, with normal macroscopic findings. Duodenal mucosal biopsies showed mostly normal findings, but also a small focus with densely packed and slightly atypical mast cells that was confirmed by immunohistochemical staining (Figure 2). The presence of the KIT-D816 V mutation was confirmed in skin and blood.
The patient had an aggressive form of systemic mastocytosis. Disease-modifying therapy was indicated, mainly on account of the anaemia, which was caused by mast cell infiltration of the bone marrow. He had also recently been diagnosed with folic acid deficiency and had constitutional symptoms that were probably a result of the mastocytosis.
The patient achieved symptomatic relief with the aid of a histamine antagonist (cetirizine), an antihistaminergic inhibitor of gastric acid secretion (ranitidine) and a proton pump inhibitor (omeprazole). He also received treatment with midostaurin, a kinase inhibitor that additionally inhibits KIT. Although the patient’s disease initially stabilised, after seven months he deteriorated with treatment-requiring anaemia, ascites and weight loss. He therefore received six cycles of cladribine, and responded with a reduced need for erythrocyte transfusions, stabilisation of bodyweight, less pronounced ascites and a fall in tryptase levels.
The patient has now been followed up for eight months without further treatment. He was hospitalised once for an infection with unknown focus, and underwent ascitic drainage and erythrocyte transfusion. He otherwise lives independently at home, fulfils the criteria for WHO functional class 1 and currently has no notable symptoms related to his systemic mastocytosis.
Discussion
Systemic mastocytosis was first described in France in the 1930s (6). Its prevalence is uncertain (7), although a retrospective cohort study of adults diagnosed with systemic mastocytosis in Denmark showed an annual incidence of 0.89 per 100 000 (8). Patient registries are now being established to obtain accurate data on incidence and prevalence (9).
An interdisciplinary approach is often necessary to achieve a correct diagnosis in patients with complex clinical pictures, such as the current patient. The signs and symptoms of mastocytosis result from a combination of mast cell activation with release of mast cell mediators, and mast cell infiltration of organs. The diagnosis is confirmed by biopsy and genetic analysis of the KIT mutation. Tryptase, an enzyme found in mast cell granules, can also provide diagnostic information and can be used in follow-up, as increasing tryptase levels are associated with a greater mast cell burden and disease progression (10).
There are three variants of mastocytosis: mast cell sarcoma, cutaneous mastocytosis and systemic mastocytosis (9). Mast cell sarcoma is a very rare and malignant form of solid tumour with the capacity for destructive infiltration and metastasis. In cutaneous mastocytosis, the pathological mast cells are restricted to the skin and give rise to various types of rash, the most common being the maculopapular rash urticaria pigmentosa (9). When the disease is identified in another organ besides the skin, it is defined as systemic if the criteria in figure 3 are fulfilled (11). Our patient fulfilled the major criterion and three minor criteria. Bone marrow is the most commonly affected site (6), but mast cell infiltration can also occur in the liver, spleen, gastrointestinal tract, skeleton and other organ systems. The prevalence in these different organs is unknown (9). Our patient had no signs of skeletal involvement on CT. His initial back pain was probably unrelated to the mastocytosis.
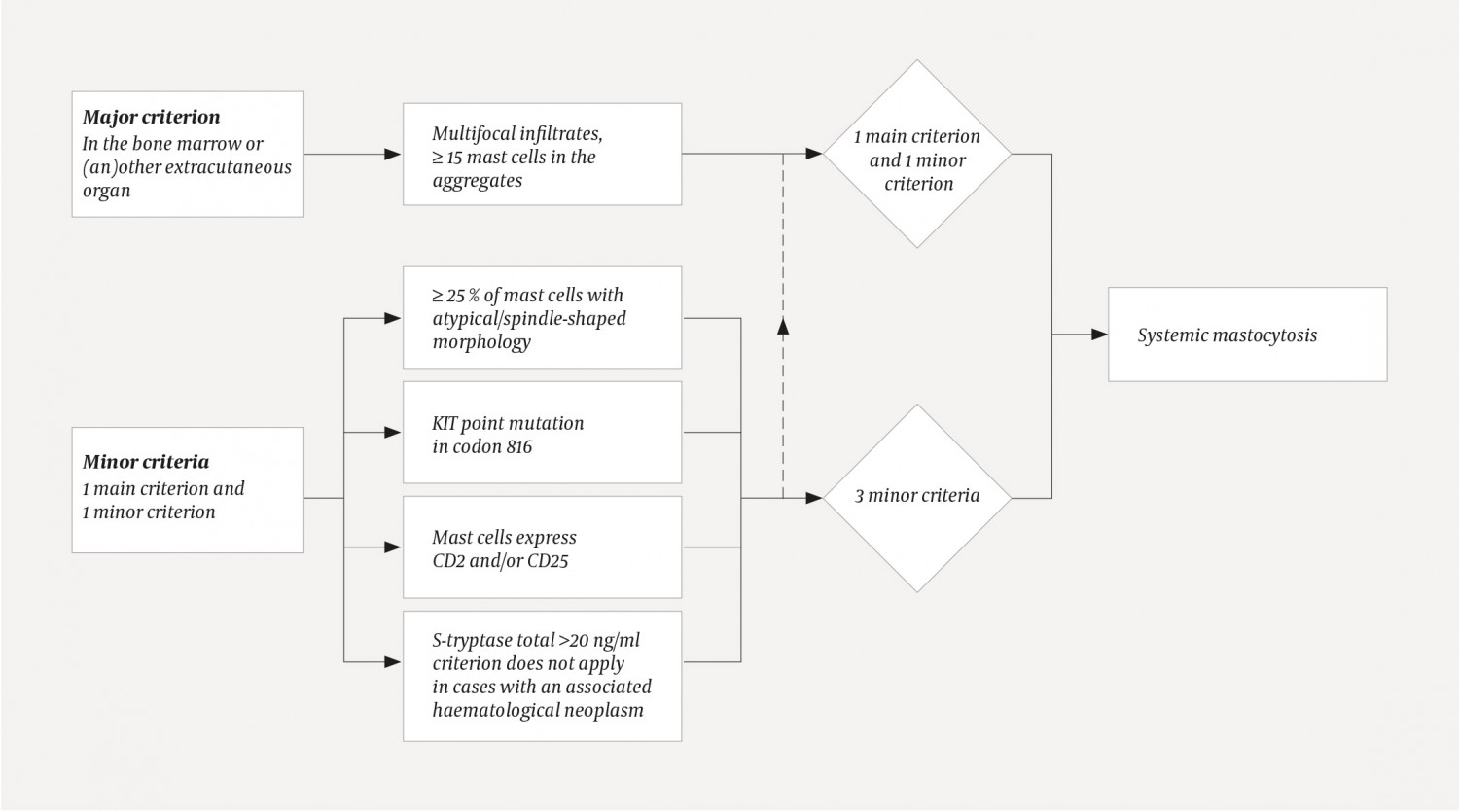
Figure 3 Diagnostic algorithm for systemic mastocytosis. A diagnosis requires one major criterion plus ≥1 minor criterion/a or ≥3 minor criteria. The criteria were defined by the World Health Organization in 2001 and were last updated in 2016 (11).
Mastocytosis is thought to originate from mutated haematopoietic stem cells (12). Studies have shown that >90 % of patients have an activating mutation in the KIT gene, of whom >80 % have a specific point mutation in exon 17, KITD816 V (13, 14). KIT is a transmembrane receptor tyrosine kinase for stem cell factor CD117 (6, 13). Mutation of KIT results in ligand-independent constitutive activation and autophosphorylation of tyrosine kinase (15) and stimulates proliferation and survival of mast cells (16).
Systemic mastocytosis was formerly classified as a myeloproliferative neoplasm (17). In accordance with the WHO classification from 2016, the condition is now considered a separate disease entity owing to unique clinical and pathological features and a highly variable disease course (18). Indolent systemic mastocytosis has a good prognosis with almost normal life expectancy (9). Clinical findings that are indicative of organ damage caused by mast cell infiltration are referred to as ‘C findings’ (9, 19). Systemic mastocytosis is defined as ‘aggressive’ in the presence of at least one C finding; aggressive systemic mastocytosis has a poorer prognosis (10).
For indolent systemic mastocytosis, treatment is mainly focused on symptom management and monitoring (6). This is based primarily on the use of antihistamines, antihistaminergic gastric acid secretion inhibitors, proton pump inhibitors, leukotriene receptor antagonists, cromoglycate and possibly steroids (6, 9, 20, 21). Patients with systemic mastocytosis are also at risk of episodes of pronounced mast cell activation with the release of large amounts of histamine and other inflammatory markers. Triggers can be emotional stress, trauma, infections, allergic reactions and medications such as acetylsalicylic acid, non-steroidal anti-inflammatory drugs (NSAIDs), opiates, anticholinergics and some contrast agents (6, 21).
Our patient was assessed as having aggressive mastocytosis, although it was difficult to determine the extent to which his associated haematological neoplasm – in the form of unclassifiable myeloproliferative disease – contributed to the clinical picture. There is a general consensus that systemic mastocytosis and any associated haematological disease should be treated independently of one another (9). Treatment of aggressive systemic mastocytosis with disease-modifying therapy remains a challenge. Some patients respond to cladribine or interferon, but most relapse or prove to have resistant disease (20). A response rate of 50–55 % to cladribine was seen in 22 patients with advanced mastocytosis. A very good (major) response, defined as the complete absence of at least one previous form of mastocytosis-related organ damage, was achieved in 37 % (22).
The tyrosine kinase inhibitor midostaurin is effective in inhibiting activated KIT (23). However, even if initial disease control is achieved, this does not always result in lasting remission. A study with midostaurin in 116 patients with advanced systemic mastocytosis showed an overall response rate of 60 %, with a very good response in 45 % of patients (23). The drug was not tested against any other form of treatment, and its effect was temporary in the majority of patients, with median progression-free survival of 14.1 months (23). Other drugs, including alternative KIT inhibitors, are undergoing testing in various phases of clinical trials, with a view to achieving disease control (6). Stem cell transplantation should be considered for young patients with aggressive disease, as it is the only treatment that offers the possibility of a lasting response and cure (6, 9). Genetic analyses with next-generation sequencing may contribute to better prognostic evaluation and more personalised treatment for patients with systemic mastocytosis (6, 24, 25).
Systemic mastocytosis can affect many organ systems. It should be considered a differential diagnosis in particular in cases of diffuse symptoms in combination with skin rash. The disease should be suspected in cases of unusual, repeated or episodic anaphylactoid reactions, flushing and syncope. The skin of such patients should be inspected for the characteristic rash urticaria pigmentosa (cutaneous mast cell infiltration). Elevated tryptase levels and possible eosinophilia strengthen suspicion of systemic mastocytosis (26). Due to the risk of anaphylactoid reactions, it is recommended that all patients with systemic mastocytosis carry adrenaline pens, and healthcare personnel must be aware of the risk associated with this patient group.
