A primigravida was found to have anaemia, leukopenia and thrombocytopenia. A broad exploratory assessment yielded no diagnostic explanation. In the course of her pregnancy she required frequent transfusions of erythrocytes and platelets, up to three times a week. The final diagnosis was not made until after the birth.
A woman in her mid-20s was referred by her local hospital to a haematologist for assessment of pancytopenia. She had been assessed two years earlier following an incidental discovery of thrombocytopenia without the cause being found. She was now primigravida in week 12. Blood tests showed: haemoglobin 8.4 g/dl (11.5–16.0 g/dl), platelets 24 · 109/l (150–450 · 109/l) and leukocytes 3.7 · 109/l (4.0–11.0 · 109/l) with neutrophil granulocytes 1.5 · 109/l (1.6–8.3 · 109/l). The reticulocyte count was normal (47 · 109/l (28–99 · 109/l)). The anaemia was macrocytic (MCV 111 fl (29.7–36.6 fl)).
Many differential diagnoses are possible in cases of pancytopenia (1, 2). Some are life-threatening, such as acute leukaemia or other malignant infiltration of the bone marrow. The combination of macrocytic anaemia and pancytopenia can be seen in cases of vitamin B12 or folate deficiency, medication use, abuse of alcohol, liver disease, myelodysplastic syndrome and aplastic anaemia. In myelodysplastic syndrome there is ineffective, dysplastic haematopoiesis and typically a cell-rich bone marrow, while aplastic anaemia is characterised by a deficiency of cells in the bone marrow. Splenomegaly with hypersplenism, viral infections and autoimmune diseases can also cause pancytopenia.
The patient reported distressing gum bleeding during the pregnancy, and that she easily bruised. She did not have fever, night sweats, or weight loss and had not recently suffered an infection. She used no regular medication except 0.4 mg folic acid supplement and did not drink alcohol. A clinical examination showed her to be in good general condition, at 153 cm in height and with a BMI above average. She had slight gingivitis in the lower jaw and several hyperpigmented macules on her skin. There were no petechiae or enlarged lymph nodes. Liver and spleen were not palpable.
Blood tests did not reveal folic acid or vitamin B12 deficiency. Liver tests and immunoglobulin level were normal. Virus serology for cytomegalovirus, Epstein-Barr virus, HIV and hepatitis C were negative. Hepatitis B antibodies in serum were elevated after earlier vaccination. No nuclear or platelet antibodies were detected. A peripheral blood smear contained no blasts, which made acute leukaemia less likely but did not exclude it. A crista biopsy and bone marrow aspirate were taken for smears, immunophenotyping and cytogenetic testing. Morphologic study and immunophenotyping of the bone marrow provided no evidence of acute leukaemia, other malignancy or myelodysplastic syndrome. The bone marrow smear revealed a reduced proportion of megakaryocytes, and contained fewer cells than expected given her age. This could be due to inmix of blood, or a genuine deficiency of cells in the bone marrow consistent with aplastic anaemia. However, the crista biopsy revealed cell-rich bone marrow (cellularity 80 %). Review of previous blood test results from several hospitals showed asymptomatic thrombocytopenia (50–80 · 109/l) in the patient since the age of 17. Two previous crista biopsies were described as normal.
The patient was discharged with a recommendation of weekly blood tests and platelet transfusions in order to maintain her thrombocyte level >25–30 · 109/l because of the increased bleeding tendency at a lower level. Blood transfusions were also recommended at haemoglobin < 8 g/dl. She needed weekly platelet transfusions from the time of her discharge. Because of the long travel distance to the hospital, the transfusions were mainly carried out at the local municipal nursing home, in agreement with her GP and haematologist. She was given a central venous catheter to facilitate blood sampling and transfusions.
The clinical situation was challenging, with severe transfusion-demanding pancytopenia of unknown cause during pregnancy. We suspected aplastic anaemia, but this diagnosis presupposes a hypocellular bone marrow in a crista biopsy, whereas the patient had a hypercellular bone marrow. In aplastic anaemia, areas of vigorous haematopoiesis can occur among areas with a paucity of cells. Thus, more biopsies may have clarified the situation, but we did not want to repeat biopsies during her pregnancy. Cytogenetic studies of the bone marrow can detect abnormalities that point to, for example, myelodysplastic syndrome, but these abnormalities are less common in aplastic anaemia. The patient had a normal bone marrow karyotype (46.XX). In aplastic anaemia (20 %), but also in some cases of myelodysplastic syndrome (1–2 %) and other bone marrow failure syndromes, cell clones associated with paroxysmal nocturnal haemoglobinuria can be detected (3). No such cell clones were detected in our patient. The combination of thrombocytopenia from her teens, short stature and several hyperpigmented cutaneous macules aroused suspicion of a hereditary bone marrow failure syndrome.
A blood sample was sent for sequencing of a gene panel for hereditary bone marrow failure. During the consultation with a medical geneticist, it emerged that the patient’s parents were remotely related and that the patient and her partner were second cousins. There were no cases of bone marrow failure in the family. She had normal facial features and normal cognitive function. She had short stature (1 percentile) and was relatively macrocephalic (90 percentile). She had many freckles on her face, 4–5 café-au-lait macules > 1.5 cm on her body and freckles and hyperpigmentation in her armpits (Figure 1). Her hands, particularly her thumbs, and nails were unremarkable.

Figure 1 The patient had hyperpigmentation and freckles in her armpits.
One diagnosis we considered was Fanconi anaemia. This disease classically presents in childhood with bone marrow failure combined with typical clinical findings, particularly skeletal anomalies of the thumb or radius and microcephaly (4). This did not fit our patient. Short stature, café-au-lait macules and freckles in the armpits can be seen with Fanconi anaemia, but are also typical of neurofibromatosis type 1, as is relative macrocephaly. However, neurofibromatosis type 1 could not explain the bone marrow failure, and sequencing (cDNA-based) of the NF1 gene associated with this condition was normal. We awaited results from the gene panel for hereditary bone marrow failure, having particularly in mind recessive conditions due to the relationship between the woman’s parents. Both parents gave blood samples in order to facilitate the interpretation of any findings of gene variants of uncertain significance, and in order to determine inheritance.
During the course of pregnancy, the need for transfusion of platelets and erythrocytes increased. Towards the end of pregnancy she needed platelet transfusions three times a week. We feared the development of platelet antibodies, resulting in less effective transfusions, but her platelet count rose adequately after the transfusions throughout the pregnancy. The number of neutrophil granulocytes gradually decreased to 1.0 · 109/l (1.6–8.3 · 109/l) without any tendency to infection. Gestational diabetes detected in week 25 was well regulated through dietary adjustments. Regular ultrasound scans showed normal fetal growth (upper percentile) and development.
Hospitalisation was planned from week 37 for monitoring and induction of labour because of a high pre-pregnancy BMI of 34 kg/m2, maternal height of 153 cm and a relatively large fetus. The hospitalisation was postponed due to the patient’s wish. In week 38 she was admitted acutely to the labour ward because of suspected severe preeclampsia with high blood pressure, proteinuria, headache and visual disturbances. Abdominal ultrasound and fetal CTG were normal. Prior to the planned induction, she had sudden onset of a strong headache, tremor and rising blood pressure, interpreted as threatening eclampsia. This prompted an acute caesarean section, with magnesium sulphate as seizure prophylaxis. Two platelet units were administered preoperatively to ensure platelets >50 · 109/l. In addition, tranexamic acid was administered intravenously. A healthy baby of normal weight was delivered. Severe uterine atony occurred peroperatively, and the patient lost a total of 2 500 ml blood. She therefore received six units of plasma, five SAG and one platelet concentrate. A Bakri balloon was also inserted in the uterus as a tamponade against the haemorrhage. Apart from a urinary tract infection, the post-operative course was otherwise uncomplicated.
A week after birth, we received the gene panel results, which pointed to Fanconi anaemia. Two previously published and assumed pathogenic variants were found in the FANCA gene (5, 6). One was an apparently synonymous (“silent”) variant (NM 000135.3:c.3624C>T p. (Ser1208=) inherited from the father. The other FANCA variant (NM_000135.3:c.3829–9G>A) could potentially affect gene splicing, but there was no evidence to confirm aberrant splicing in the literature. Major gene deletions are not usually detected by sequencing, and for the sake of good order a manual review of the sequencing data was performed with this in mind. The FANCA allele inherited from the mother proved to contain both c.3829–9G>A and an exon 1–12 deletion. The latter is pathogenic, while c.3829–9G>A was regarded as probably not pathogenic per se.
The deletion, together with the paternally inherited, pathogenic variant, resulted in “compound heterozygosity”, which confirmed the clinical suspicion of autosomal recessive Fanconi anaemia. The parents were asymptomatic carriers of different pathogenic gene variants, and the familial relationship between them was therefore irrelevant.
Testing of mRNA confirmed that the gene copy with c.3829–9G>A and exon 1–12 deletion could not produce protein. The allele inherited from the father resulted partly in normal mRNA and partly in aberrantly spliced mRNA with a premature stop codon (p.Ser1208Ilefs*28). Production of some normal mRNA from the gene copy with the “milder” synonymous variant may help to explain the late onset of illness in the patient.
Testing was supplemented by a chromosomal breakage analysis, which measures the capacity for DNA repair in lymphocytes after exposure to DNA cross-linking agents. The test has high sensitivity and specificity for Fanconi anaemia when a large number of chromosome breaks are found. Our patient had a large number of chromosome breaks, but somewhat fewer than in classic Fanconi anaemia. This was consistent with our patient having a milder variant of the disease.
Fanconi anaemia is a rare genetic condition with a prevalence previously estimated at 1–9/1 000 000 (7). Pathogenic mutations have been found in 21 different genes, most frequently the FANCA gene. Inheritance is usually autosomal recessive. The disease is due to impaired ability to repair DNA damage, which may cause varying degrees of bone marrow failure. Fanconi anaemia also entails a substantially increased risk of cancer, both haematological, such as acute myelogenous leukaemia and myelodysplastic syndrome, as well as solid tumours, particularly squamous cell carcinomas (4). There is typically also a combination of skeletal anomalies, café-au-lait macules, growth retardation, congenital malformations and microcephaly (4). The disease is usually diagnosed in childhood. Only 10 % are over the age of 16 at the time of diagnosis (8).
A further crista biopsy after the birth (Fig. 2) confirmed hypocellular bone marrow (cellularity 15–20 %), consistent with aplastic anaemia, a known complication of Fanconi anaemia.
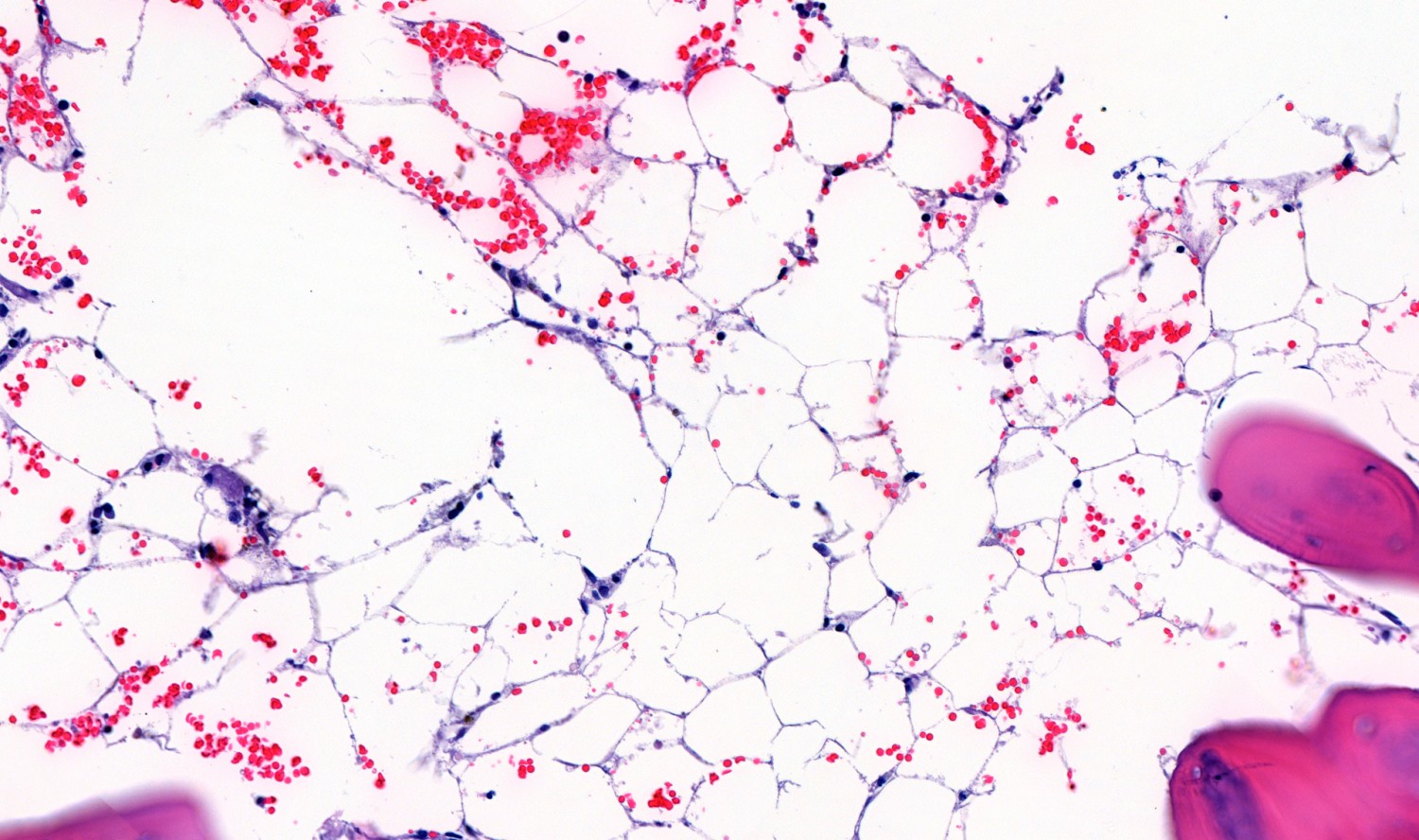
Figure 2 Detailed image of crista biopsy showing acellular area replaced by marrow adipose tissue.
For a diagnosis of aplastic anaemia, a hypocellular bone marrow is required (<25 % cellularity in crista biopsy). At least two of the following criteria must also be met: haemoglobin < 10 g/dl, platelet count < 50 · 109/l and neutrophil granulocytes < 1.5 · 109/l (9). 70–80 % of aplastic anaemia cases are idiopathic, while the remainder are primarily hereditary bone marrow failure syndromes (9). Fanconi anaemia is the most common of these (8).
The patient was told that allogeneic bone marrow transplantation might be indicated. Her pancytopenia gradually improved. Two months after giving birth, she was assessed for allogeneic bone marrow transplantation with an unrelated donor, but it was decided to wait, as her bone marrow failure was stable without need for transfusions.
Allogeneic bone marrow transplantation is the only curative treatment for severe bone marrow failure in Fanconi anaemia, but is associated with high mortality and morbidity. The treatment does not reduce the general risk of cancer other than haematological malignancy, and also increases the risk of solid tumours in this patient group (10).
One year after delivery the patient was still transfusion independent and was closely monitored by a cross-disciplinary team. Her haemoglobin level was stable at 8–9 g/dl, neutrophil granulocytes at 0.5–1.5 · 109/l and blood platelet count 20–35 · 109/l. Because the patient was related to her partner, we performed carrier testing of the FANCA gene in him. Normal findings in the partner meant that we did not need to examine the healthy child.
Discussion
This case report illustrates the fact that Fanconi anaemia is a possible diagnosis also in adulthood in cases of isolated cytopenia or pancytopenia. Besides bone marrow failure, our patient had only modest clinical signs pointing to Fanconi anaemia in the form of slightly short stature and café-au-lait macules. Freckles in the armpits are a rare and characteristic finding, but not pathognomonic for the condition. It has been reported that 25 % of patients do not have physical anomalies (4). Around 90 % have a varying degree of bone marrow affectation by the age of 40 years (10). Thrombocytopenia tends to occur first, and over 90 % of patients have an elevated mean corpuscular volume (MCV) (11), as in the case of our patient. In a few cases, the diagnosis may also be made in adults who are found to have cancer types associated with Fanconi anaemia. Given greater attention to milder/later onset forms, and more readily available genetic diagnostics, it is probable that the prevalence estimate will increase. Studies of carrier frequencies also support this (12).
It is important to diagnose Fanconi anaemia at an early age because cancer screening and advice on cancer prophylaxis are indicated. Patients should be checked regularly for the development of myelodysplastic syndrome, acute myelogenous leukaemia and squamous cell carcinomas of the head and neck region, vulva, vagina and cervix (4). Radiological imaging must be avoided as far as possible, as radiation increases the risk of cancer. If a cancer diagnosis is made, Fanconi anaemia has a bearing on the choice of therapy, as these patients have lower tolerance for chemotherapy and radiotherapy (4). Pregnant women with Fanconi anaemia require close monitoring. Transitory severe bone marrow failure during pregnancy has been described previously (13, 14). This is due to increased haematopoietic requirements in pregnancy combined with an impaired bone marrow response (13). Increased risk of preeclampsia, eclampsia and caesarean sections has also been reported previously (13, 14).
Fanconi anaemia is a possible diagnosis in young adults < 40–50 years with aplastic anaemia (9) or myelodysplastic syndrome (15). This case history illustrates that the diagnosis should also be considered for young people with bone marrow failure of unknown etiology, as our patient had repeated crista biopsies showing normal cellularity before the final biopsy revealed aplastic anaemia. When Fanconi anaemia is suspected, we recommend that the patient should be referred to a haematologist and that both a chromosomal breakage analysis and a gene sequencing panel for hereditary bone marrow failure are performed. If the chromosomal breakage analysis is normal, larger gene panels could potentially detect another hereditary cause of bone marrow failure. When the results of the chromosomal breakage analysis are aberrant, but no definite pathogenic variants are detected by the gene sequencing panel, examination for deletions in the FANCA gene is important.
