A middle-aged man was admitted with elevated liver values and jaundice. Because malignancy was suspected, the man underwent extensive liver surgery. Histological examinations of the liver resection specimen revealed an unexpected and underdiagnosed disease.
A man in his fifties with a history of gout presented to his GP reporting two months of central abdominal pain. His prescribed medications included colchicine tablets (0.5 mg) as needed. He had never smoked and his alcohol consumption was insignificant. Preliminary blood tests showed slightly elevated amylase at 168 U/l (reference range 10–65 U/l). Liver transaminases, bilirubin and alkaline phosphatase were normal. Abdominal ultrasound and magnetic resonance cholangiopancreatography (MRCP) revealed normal liver and bile duct findings, but the pancreas was atrophic with an irregular pancreatic duct consistent with chronic pancreatitis.
The patient was referred to the Gastrointestinal Surgery Outpatient Clinic for further assessment. At this consultation, which took place eight weeks later, blood samples showed normal values of liver transaminases and alkaline phosphatase, and an almost normalised amylase value of 68 U/l (10–65 U/l). The gall bladder could not be assessed in previous diagnostic imaging. Transabdominal ultrasound was therefore repeated and gallstone disease was excluded. The patient’s diagnostic imaging findings were not typical of autoimmune pancreatitis, where one normally sees homogeneous enhancement in an oedematous and enlarged pancreas. Autoimmune pancreatitis was therefore not considered as a differential diagnosis at this point.
The most common causes of chronic pancreatitis are alcohol consumption and smoking, and a thorough clinical history is therefore important. Obstruction of the pancreatic duct due to gallstones, strictures or tumours must be excluded (1). Autoimmune pancreatitis (AIP) is a differential diagnosis that it is important to identify because it responds well to steroid treatment. Autoimmune pancreatitis is divided into types 1 and 2. Type 1 AIP is regarded as a manifestation of IgG4-related disease and serum IgG4 (immunoglobulin G subclass 4) is elevated. In type 2 AIP serum IgG4 is normal (2). Hyperlipidaemias, hypercalcaemia, chronic renal failure and drugs are other causes of chronic pancreatitis. Pancreatitis may also be congenital, and it is important to determine this because patients with hereditary pancreatitis have a higher risk of adenocarcinoma in the pancreas. The autosomal dominant PRSS1 mutation, in particular, increases risk. Genetic guidance is recommended for patients under 25 years of age with new-onset idiopathic pancreatitis and for those who have several first-degree relatives with chronic pancreatitis (1). A review article on chronic pancreatitis was published in the Journal of the Norwegian Medical Association in 2018 (1).
The patient consulted his GP again six weeks after the consultation at the Gastrointestinal Surgery Outpatient Clinic. He had had a gradual weight loss of 16 kg, from 83 to 67 kg, over the previous six months. During the last few weeks he had developed diarrhoea, with 10–15 bowel movements per day. He had noticed greyish faeces that floated in the toilet and dark urine. He had also experienced enlarged submandibular nodes and for the past week he complained of severe pruritus which impacted his sleep. He did not have night sweats, and by this time the abdominal pain had ceased. His GP took new blood samples, and liver test results were now found to be abnormal, with gamma-glutamyl transferase (GGT) 1 170 U/l (15–115 U/l), alkaline phosphatase 243 U/l (35–105 U/l), alanine aminotransferase (ALT) 380 U/l (10–70 U/l) and bilirubin 28 µmol/l (< 25 µmol/l). Laboratory tests showed leukocytes 4 ∙ 109/l (3.5–8.8 ∙ 109/l), sedimentation rate 15 mm/h (< 20 mm/h) and CRP 6 mg/l (< 5 mg/l). On suspicion of an unidentified malignant disease, the patient was referred to the Department of Internal Medicine and a standardised cancer package pathway.
The patient came for an outpatient consultation the day after receiving the referral. Eight months had now passed since the onset of symptoms. Vital signs showed blood pressure 150/105 mm Hg and pulse rate 73/min. The patient had lymphadenopathy with bilateral enlargement of submandibular glands measuring about 4x 3 cm and a small lymph node in the left groin which was regarded as probably pathological. His general condition appeared slightly reduced. Computed tomography (CT) scan of collum, thorax, abdomen and pelvis revealed a diffuse mass lesion in the liver hilum growing into the common hepatic duct (Fig. 1a). The swollen glands on his neck were described as prominent submandibular salivary glands, and were not suspicious for metastasis (Fig. 1b). The pancreas was atrophic as in the previous MRI examination (Fig. 1c). The finding aroused strong suspicion of perihilar cholangiocarcinoma.
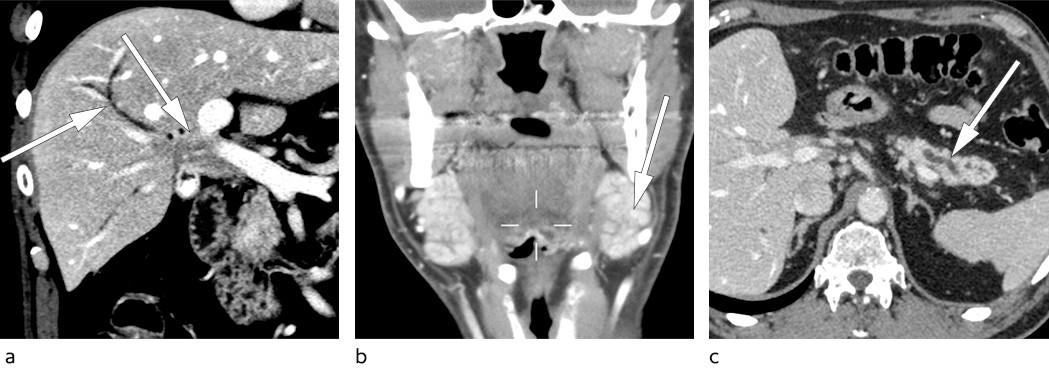
Figure 1 a) CT abdomen showing a mass lesion in the liver hilum with dilated intrahepatic bile ducts. b) CT collum showing bilateral enlargement of the submandibular glands (so-called Küttner tumour). c) CT abdomen with finding of atrophic pancreas with dilated and irregular pancreatic duct.
Cholangiocarcinoma may occur along any part of the biliary tract and is a rare tumour in the Western world with an incidence of about 1–2 per 100 000 (3). Primary sclerosing cholangitis is the main risk factor, but most cholangiocarcinomas arise spontaneously. Perihilar cholangiocarcinoma is the most common type, developing at or near the confluence of right and left hepatic ducts(3). They are also called Klatskin tumours, after Dr Gerald Klatskin, who described the condition more than 50 years ago (4). The diagnosis is usually made late, and 90 % of patients present with silent jaundice. The prognosis for cholangiocarcinoma is very poor, with a five-year survival for the entire group of less than 20 %. The only curative treatment is surgery, but only 25–30 % of the patients are resectable at the time of diagnosis (3). The tendency in recent years has been for more aggressive surgery, including liver transplantation for carefully selected patients.
The patient developed jaundice and was admitted to the Department of Gastrointestinal Surgery. Blood tests on admission showed bilirubin 130 µmol/l. An MRCP examination revealed a long occlusion at the confluence of right and left ducts with dilatation of intrahepatic bile ducts. Calibre variation consistent with primary sclerosing cholangitis was not present, and the value of the tumour marker carbohydrate antigen 19–9 (CA19–9) was normal at 7 kU/l (< 35 kU/l). High CA19–9 serum values may be found with pancreatic or bile duct cancer, but are also seen in bile duct inflammation and benign cholestasis. Low CA19–9 levels may be seen even in cases of advanced cholangiocarcinoma (3). The patient was discussed at multidisciplinary team meetings, and the findings were considered to be most compatible with cholangiocarcinoma. It was decided that the patient was suitable for surgery.
Because of a rise in bilirubin level to 190 µmol/l and intense pruritus, endoscopic retrograde cholangiopancreatography (ERCP) was performed, and a double pigtail stent placed through the biliary stricture of the liver hilum. Biliary brush cytology showed normal bile duct epithelium. Despite a well-placed stent, the patient became increasingly jaundiced and blood tests showed increasing bilirubin levels, from 190 µmol/l to 230 µmol/l. There was no clinical or biochemical suspicion of cholangitis. Percutaneous transhepatic cholangiography was used to insert a drain in the left main bile duct. In addition, ERCP was again performed and the pigtail stent was replaced with two straight plastic stents in the right and left main bile ducts. Despite this, his bilirubin concentration remained high at 200 µmol/l.
Preoperative drainage of the liver was performed on our patient to reduce the risk of post-operative liver failure. In some cases, it is necessary to combine endoscopic and percutaneous drainage. In an ERCP procedure, one or more stents may be placed beyond the malignant obstruction. Because of the risk of complications, this procedure is today performed almost exclusively in a therapeutic context (5).
Despite the well-placed stents in the bile ducts, the bilirubin value remained high. This might be due to serum delta-bilirubin, which is formed in cases of prolonged obstructive jaundice and has a half-life of two to three weeks. At this point the patient was transferred to Rikshospitalet for surgery.
Six days after the transfer, extended right-sided liver resection with hepaticojejunostomy was performed. The patient developed liver failure post-operatively, with subsequent ischaemic necrosis in residual liver tissue. His condition was judged to be very serious with little chance of spontaneous improvement. Examination of the resection specimen was therefore classified as urgent, in case the condition should prove to be benign and hence allow transplantation. A histological sample from the liver hilum showed no signs of malignancy, but lymphoplasmacytic infiltrates and abundant eosinophil granulocytes. Immunohistochemical analysis revealed an excess of IgG4-positive plasma cells and an elevated IgG4/IgG ratio of over 0.4 (Fig. 2). Serum-IgG4 concentrations were measured post-operatively, and found to be elevated at 6.0 g/l (0.03–2.01 g/l). The findings were consistent with IgG4-related disease.
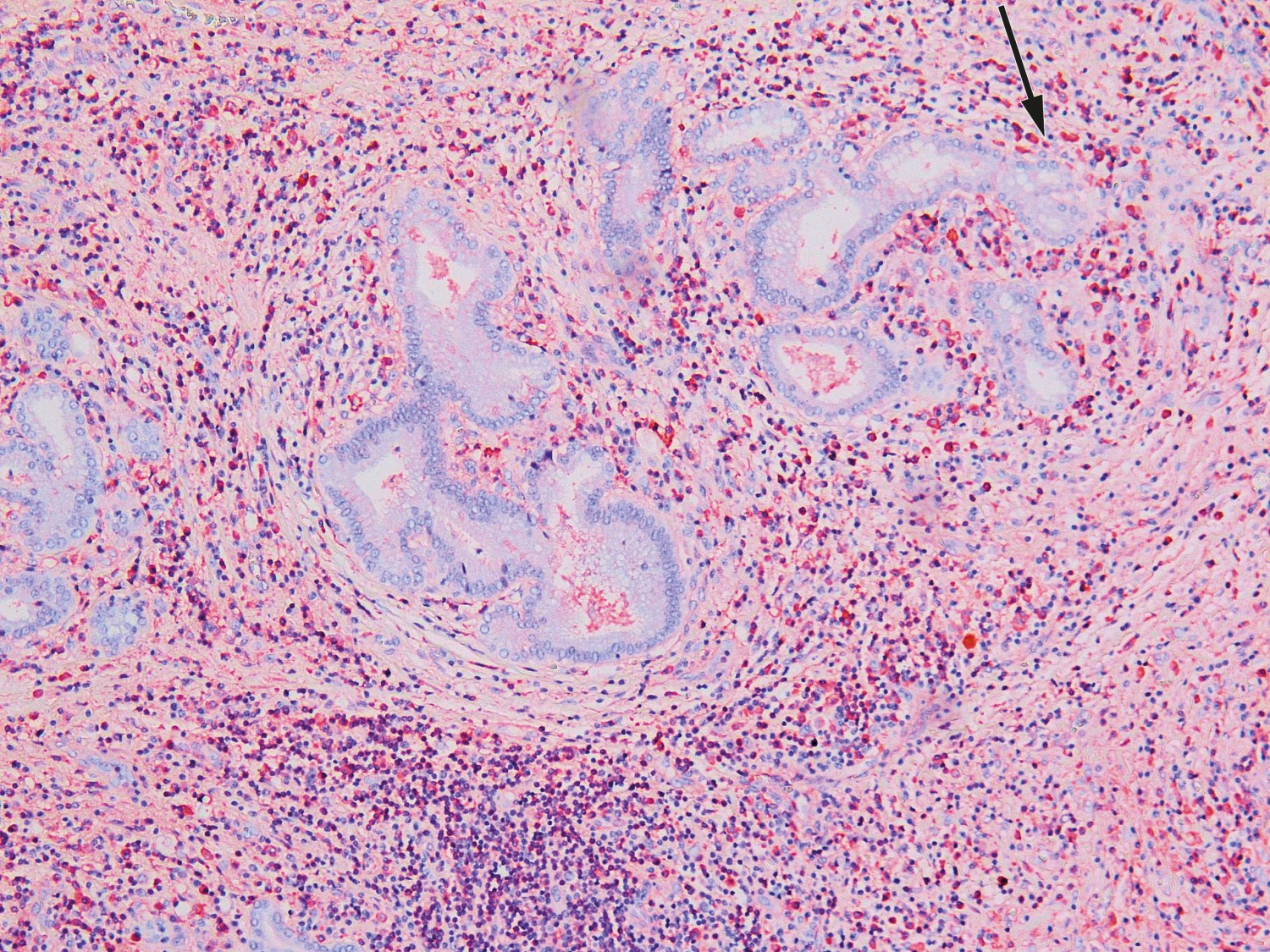
Figure 2 Histological section from liver hilum with lymphoplasmacytic inflammation and abundant eosinophil granulocytes. An immunohistochemical examination revealed IgG4-positive plasma cells (the red cells) and an IgG4/IgG ratio of over 0.4.
The patient was listed for liver transplant. In the meantime, his clinical condition deteriorated considerably, with slurred speech, disorientation and lethargy, and 14 days after the liver resection he was transferred to the Intensive Care Unit. There was a significant rise in ammonia concentration (78 μmol/l, reference range < 35 μmol/l), lactate (5.8 mmol/l, < 2.0 mmol/l), lactate dehydrogenase (659 U/l, 105–205 U/l), aspartate-amino transferase (AST) (1 232 U/l, < 45 U/l) and ALT (784 U/l). INR was 1.5 and creatinine 48 µmol/l (60–105 µmol/l). His bilirubin level remained stable at around 200 µmol/l. CRP and procalcitonin levels continued to rise at 135 mg/l and 1.9 µg/l (< 0.1 µg/l) respectively despite ongoing intravenous therapy with piperacillin and tazobactam. An abdominal CT scan showed progression of necrotic areas in the liver and increasing ascites with signs of peritonitis. Because of the clinical exacerbation with encephalopathy and considerably deteriorated general condition, the patient’s status on the waiting list for liver transplantation was changed to “urgent”. Seventeen days after the liver resection the patient underwent a liver transplantation, and three weeks later he could be discharged to his home.
The patient is now immunosuppressed with prednisolone, mycophenolate mofetil and tacrolimus. At follow-up one year after liver transplantation the patient has no palpable salivary glands, and his serum-IgG4 has normalised to 1.1 g/l (0.03–2.01 g/l). The patient is monitored regularly for signs of IgG4-related disease relapse.
Discussion
IGg4-related disease is a systemic, immune-mediated disorder characterised by inflammation and progressive fibrosis. The condition was first described in the literature in 2003, and in 2011 the concept IgG4-related disease was defined. A review article on IgG4-related disease was published in the Journal of the Norwegian Medical Association in 2017 (2).
The typical patient with IgG4-related disease is a middle-aged or elderly man. Autoimmune pancreatitis type 1 is the most common manifestation and accounts for 60 % of cases. It is followed by affection of salivary glands, kidneys, lacrimal glands and retroperitoneal fibrosis. In half of the patients, two or more organs are affected (6).
Characteristic tissue biopsy findings obtained by immunohistochemical analysis, with lymphoplasmacytic inflammation, storiform fibrosis and a ratio of IgG4- to IgG-positive plasma cells of over 0.4 are regarded as the gold standard, but the diagnosis must be based on a combination of radiological, pathological and serological findings (6). The most widely used sets of diagnostic criteria are the HISORt criteria (histology, imaging, serology, other organ involvement and response to steroid therapy), which were developed for diagnosing autoimmune pancreatitis and later modified for IgG4-related sclerosing cholangitis (7).
Serum-IgG4 levels are neither sufficiently sensitive nor specific, and have no established cut-off value (6). Elevated IgG4 may be seen in association with malignancy and other inflammatory disease, for example in 10–15 % of patients with primary sclerosing cholangitis (8). Elevated serum IgG4 must therefore be interpreted with caution. An elevated serum IgG4/IgG1 ratio (> 0.24) and IgG4/total-IgG ratio (> 0.1) is indicated to have higher specificity than serum IgG4 alone (8). The IgG4/IgG-RNA ratio has shown promising results as a more accurate marker, but is not available for clinical use (9).
The plasmablast cell count per millilitre is often elevated and may have a diagnostic value in combination with serum IgG4 (10). Serum plasmablasts are assayed today at Oslo University Hospital, Rikshospitalet. Elevated IgE and eosinophilia are common, and increased CRP and sedimentation rate may be seen. If systemic IgG4-related disease is suspected, a PET-CT scan can be performed to assess the extent of the disease and to evaluate response to treatment (6).
Patients with IgG4-related sclerosing cholangitis are known to present with jaundice, weight loss and abdominal pain, as in the case of our patient. It may be difficult to distinguish IgG4-related sclerosing cholangitis from primary sclerosing cholangitis because MRCP and ERCP findings may be completely identical. Differential diagnosis is very important because it has consequences for therapy, follow-up and prognosis (6).
Our patient was found to have IgG4-related disease affecting the pancreas, bile ducts and salivary glands. Ηis clinical history began with the detection of chronic pancreatitis, and it is probable that the patient has had recurring IgG4-related autoimmune pancreatitis, even though the diagnostic imaging results were not typical of this disease at the time of diagnosis.
The liver resection specimen revealed that the patient had an IgG4-related inflammatory pseudotumour of the liver hilum, which may explain the ineffective biliary drainage. IgG4-related inflammatory pseudotumour of the liver is rarely reported and is most frequently described in the orbit, kidney and lungs. Tissue biopsy of perihilar strictures is largely avoided because of the fear of tumour dissemination and due to the risk of pancreatitis and cholangitis (3).
The case history shows the importance of broad differential diagnosis assessment, if the map does not agree with the terrain. In our patient’s case, the combination of ineffective biliary drainage and affection of several organs directed the diagnosis towards systemic disease. Analysis of serum IgG4 concentrations was not carried out during the workup phase, but is of limited diagnostic value as a single diagnostic marker because it may also be elevated in malignancy. Normal biliary brush cytology and normal serum CA19–9 measurements cannot be emphasised due to the low sensitivity of these tests. If IgG4-related disease had been considered prior to surgery, it would have been relevant to obtain a tissue sample from the enlarged submandibular glands and attempt steroid therapy. The dilemma in this period would be the risk of the patient developing a non-resectable cholangiocarcinoma.
IgG4-related disease is believed to be underdiagnosed because of lack of awareness of the condition (6). The disease must be regarded as a differential diagnosis when malignancy in pancreas or bile duct strictures is suspected. In a study from the Netherlands, 15 % of the patients who underwent surgery for suspected perihilar cholangiocarcinoma had a benign stricture, and 42 % of them had IgG4-related disease (11).
Early treatment is important to prevent the development of fibrosis and irreversible organ damage. In 2015, an international group of experts introduced recommendations for treating IgG4-related disease (12). The first choice of treatment is peroral prednisolone (30–40 mg daily) for two to four weeks, followed by gradual tapering (13). Most patients respond well to this, but relapse during tapering and discontinuation is common, and long-term prednisolone treatment (5–7.5 mg daily) reduces the risk of recurrence (13). Practice regarding second-line treatment varies internationally. There are no prospective studies on traditional steroid-sparing drugs such as azathioprine, methotrexate and mycophenolate mofetil, and there is limited data supporting the efficacy of these drugs for IgG4-related disease (12).
Rituximab is a monoclonal antibody for the B-cell marker CD20 and has shown promising results (14). Rituximab selectively reduces the number of CD20-positive B-lymphocytes, which in turn reduces the number of circulating plasma cells that produce IgG4. This is reflected in the fact that the number of plasmablasts (a cell stage between mature B-lymphocytes and differentiated plasma cells) per millilitre is significantly reduced after rituximab therapy (6). Treatment with rituximab in the form of two 1000 mg intravenous infusions separated by 2 weeks has proved effective in inducing remission, and the drug is usually tolerated very well (14). Clinical experience indicates that rituximab should be offered to patients who do not tolerate prednisolone, or for whom it does not have the desired efficacy. Documentation of rituximab maintenance therapy is scarce (14, 15). The pathogenesis of IgG4-related disease is only partly defined, but the good response to rituximab indicates that B-lymphocytes play a central part.
Conclusion
IgG4-related disease can mimic several malignant and inflammatory conditions, and the diagnostic workup can be difficult. When malignancy in the pancreas or biliary tree is suspected, IgG4-related disease is an important differential diagnosis, and other organ involvement must be excluded. The disorder may affect virtually any organ in the body, and knowledge of this diagnosis is relevant to all subspecialties.
