A girl of early primary school age was admitted to the paediatric department after an acute onset of dizziness, numbness in the right arm, and word-finding difficulties. The symptoms had abated upon admission, but the girl would nevertheless prove to be seriously ill.
The girl had a history of constipation and reactive airway disease, and was being monitored by a dentist because of hypomineralised tooth enamel. She was also extremely afraid of healthcare workers, which her parents suspected was the result of previous attempts to take blood samples. Ten weeks prior to the episode in question, she had had a cold that her parents described as unusually severe. She had never quite fully recovered and was still lethargic and needed a great deal of rest. The family had visited their general practitioner, but the girl’s anxiety regarding healthcare workers meant that two attempts to take blood samples both had to be aborted.
On the day of her admission, the girl experienced an acute onset of word-finding difficulties, numbness in her right arm, and dizziness so severe that she fell over while playing. The symptoms abated, but a short while later the word-finding difficulty returned. When she presented at Paediatric Acute Admissions 2.5 hours after symptom onset, her symptoms had resolved completely. She was walking around, chatting, eating and drinking, but was very tired and just wanted to sleep. Her anxiety prevented a full examination; she turned away, lay on her stomach and refused to cooperate. Vital parameters and auscultation of the heart, lungs and abdomen were normal. She was difficult to assess neurologically owing to her lack of cooperation, but showed normal and symmetrical facial expressions, pupillary reactions and movements of the extremities. An attempt was made at midazolam sedation to enable blood samples to be taken, but she resisted all attempts at medication. She found the situation stressful, began to hyperventilate and refused to listen to anyone, including her parents, whenever a healthcare worker came near her.
A number of differential diagnoses were considered at this time. The resolution of symptoms and the absence of any clear clinical findings meant that infarction, haemorrhage or a tumour were considered unlikely, even though cerebral events such as these may have a fluctuating course. A transient ischaemic attack or epilepsy were both possibilities given the duration and resolution of the symptoms. Another cause of neurological symptoms in children is hypoglycaemia. However, investigating this further would have required coercion, which, in our view, might have had the unfortunate effect of making the situation worse for the girl. While the situation was unresolved, the patient appeared to be stable. She was therefore admitted for overnight observation with further assessment planned for the following day.
When the parents went to wake the girl in the morning, she was noticeably pale and clammy, as well as unresponsive. She did not react to painful stimuli and had gurgling breath sounds, but her oxygen saturation and pulse were normal. Her pupils were dilated, but reactive to light. An alarm was raised to summon additional personnel. Shortly afterwards, the patient experienced a generalised tonic-clonic seizure that was treated with buccal midazolam.
Causes of seizures in children can be classified as structural, genetic, infectious, metabolic, immunological or idiopathic (1). Febrile seizures are a common reason for presentations at Paediatric Acute Admissions, but our patient had no signs of infection, and febrile seizures are most often seen in children below the age of five years (2). On account of the seizure and the symptoms the day before, it was now considered essential to exclude structural causes such as a tumour, haemorrhaging or infarction, as well as infection, electrolyte imbalance or a metabolic disorder.
In the postictal phase, capillary blood glucose was measured at 1.7 mmol/l (reference range 4.0–6.3 mmol/l), and treatment with intravenous glucose was immediately initiated to correct the hypoglycaemia. The patient was placed under general anaesthesia for an acute brain MRI and collection of blood samples. The MRI showed cerebral vein asymmetry, which could reflect normal anatomical variation, or possibly hyperaemia after a seizure. The MRI was otherwise normal.
In addition to the hypoglycaemia, the blood tests revealed metabolic acidosis with pH 7.10 (7.32–7.43) and bicarbonate 16 mmol/l (24–31 mmol/l), as well as electrolyte disturbances in the form of hypoosmolality (258 mosmol/kg (289–305)), hyponatraemia (119 mmol/l (137–145)), hypocalcaemia (free calcium 1.01 mmol/l (1.18–1.32)) and hyperphosphataemia (3.00 mmol/l (1.00–1.80)). The patient was transferred to the paediatric intensive care unit, where correction of electrolytes was started using intravenous fluids with additives in accordance with the emergency care guidelines of the Norwegian Paediatric Society (Akuttveileder i pediatri) (3, 4).
The brain MRI ruled out structural causes, whereas the pronounced abnormalities in electrolyte levels could explain her symptoms. Hypoglycaemia, hyponatraemia and hypocalcaemia can all trigger seizures (5). It is especially important to investigate calcium in patients with numbness and seizure tendency. The patient appeared to be euvolaemic. Syndrome of inappropriate antidiuretic hormone secretion (SIADH syndrome), in which the secretion of antidiuretic hormone is increased, glucocorticoid deficiency, stress or hypothyroidism were therefore regarded as possible explanations for the electrolyte disturbances. Of these, adrenal insufficiency was the most likely, and could also explain the acidosis. However, she was neither hypotensive nor hyperkalaemic.
The anaesthesia was discontinued, but the patient remained noticeably lethargic. Further blood test results revealed low morning cortisol at 6 nmol/l (133–537), confirming the suspected adrenal insufficiency. It was noted that she also had general hyperpigmentation, although not in the axillae or mucous membranes. In accordance with the emergency care guidelines, treatment was started with hydrocortisone 100 mg intravenously, followed by 100 mg intravenously over the next 24 hours (6). Over the course of a few hours, the patient gradually became increasingly alert, and by the next day she was back to her usual self.
Hyperpigmentation is a characteristic finding in cases of primary adrenal insufficiency because adrenocorticotrophic hormone (ACTH) also stimulates the production of melanin. This proved to be the case in our patient, whose ACTH level was 228 pmol/l (1.1–10.2). The patient’s adrenal insufficiency could explain the acidosis, hyponatraemia and hypoglycaemia, but not the hyperphosphataemia or hypocalcaemia. The calcium and phosphate balance is controlled to a large degree by parathyroid hormone (PTH). PTH increases extracellular calcium levels, and is the most important regulator of the calcium level. It also helps regulate phosphate levels by increasing renal excretion. Our patient was found to have PTH levels below the limit of quantification (<0.5 pmol/l (2.3–10.7 pmol/l)), consistent with hypoparathyroidism. This therefore explained both her hyperphosphataemia and hypocalcaemia. Acute adrenal insufficiency and hypoparathyroidism are rare diseases in children. The fact that our patient had both suggested that there may be a common underlying cause, with autoimmunity the most likely possibility.
On suspicion of autoimmune polyendocrine syndrome type 1 (APS-1), a genetic test was performed that involved sequencing the gene for the regulator AIRE. This revealed heterozygosity for two pathological variants, c.22C>T, p.(Arg8Cys) and c.1163_1164insA, p.(Met388,IlefsTer36) (Medical Genetics, Haukeland University Hospital). Further tests revealed that the patient also had anti-parietal cell antibodies (32 U/ml (0–10 U/m)), autoantibodies against intrinsic factor (357 U/ml (0–10 U/ml)), elevated anti-TPO (41 IU/ml (0–35 IU/ml)) and autoantibodies against adrenal cortex (titer > 640, positive) (Department of Immunology and Transfusion Medicine, St. Olavs Hospital). The presence of autoantibodies was further confirmed by analysing antibodies against endocrine tissue (Department of Immunology and Transfusion Medicine, Oslo University Hospital), which also contains anti-gonadal antibodies (> 100, positive). All of these findings supported the diagnosis.
During her stay in hospital, the girl had several episodes of diarrhoea. A gastroscopy was therefore performed, which revealed chronic autoimmune atrophic gastritis. She was discharged after four weeks and is now back to her usual self. She attends regular follow-up appointments and is receiving treatment with cortisone acetate, fludrocortisone, alfacalcidol (vitamin D3 preparation), calcium, and vitamin D drops. She has been referred to the Department of Endocrinology at Haukeland University Hospital, which is responsible for the Norwegian registry for organ-specific autoimmune diseases (ROAS) and has a specific focus on autoimmune polyendocrine syndrome type 1.
Discussion
Autoimmune polyendocrine syndrome type 1 is a rare disease with autosomal recessive inheritance, and has previously been described in the Journal of the Norwegian Medical Association in an adult patient (7). The syndrome is an autoimmune disorder caused by mutation of the autoimmune regulator gene AIRE (8). The AIRE gene expresses thousands of tissue-specific self-antigens in the thymus, which results in autoreactive T cells being destroyed via negative selection. A mutation in AIRE would thus enable autoreactive T cells to evade apoptosis (9). It has also been speculated that AIRE may be involved in the development of Foxp3+ regulatory T cells, which can suppress autoreactive T cells (8–10). A mutation in AIRE could thus result in autoreactive T cells avoiding destruction, and also lead to reduced formation of regulatory T cells which would normally suppress them. In patients, circulating autoantibodies and infiltrating lymphocytes are both found in affected organs, and will eventually lead to organ failure (9, 11). The prevalence of the disorder is estimated to be 1: 100 000 worldwide; in Norway it is estimated to be 1: 90 000, while in Finland and among Iranian Jews, it may be as high as 1: 25 000 and 1: 9 000, respectively (9, 12–14).
Clinically, autoimmune polyendocrine syndrome type 1 is characterised by a classic triad of chronic mucocutaneous candidiasis, hypoparathyroidism and adrenal insufficiency (15). The disease can have a variable presentation with a number of other manifestations (Figure 1) (7, 16). In a Norwegian study, 52 patients with the disease showed a median of five disease manifestations (11). It is common for the first manifestation to appear in childhood, typically mucocutaneous candidiasis, with hypoparathyroidism and adrenal insufficiency occurring later (17). However, in the Norwegian study, hypoparathyroidism was somewhat more common than chronic mucocutaneous candidiasis as the first manifestation, and 40 % of patients developed the entire triad (11). In a Finnish study, the initial presentation was mucocutaneous candidiasis, and 23 % of patients had 1–6 other manifestations before developing the classic triad (15). Enamel hypoplasia, which was also present in our patient, is an early finding in many affected individuals (11, 18). Early onset of the disease has been reported to lead to a more severe course, with a greater number of manifestations (11). The disease thus shows extensive phenotypic variation, which may lead to late detection and delayed treatment.
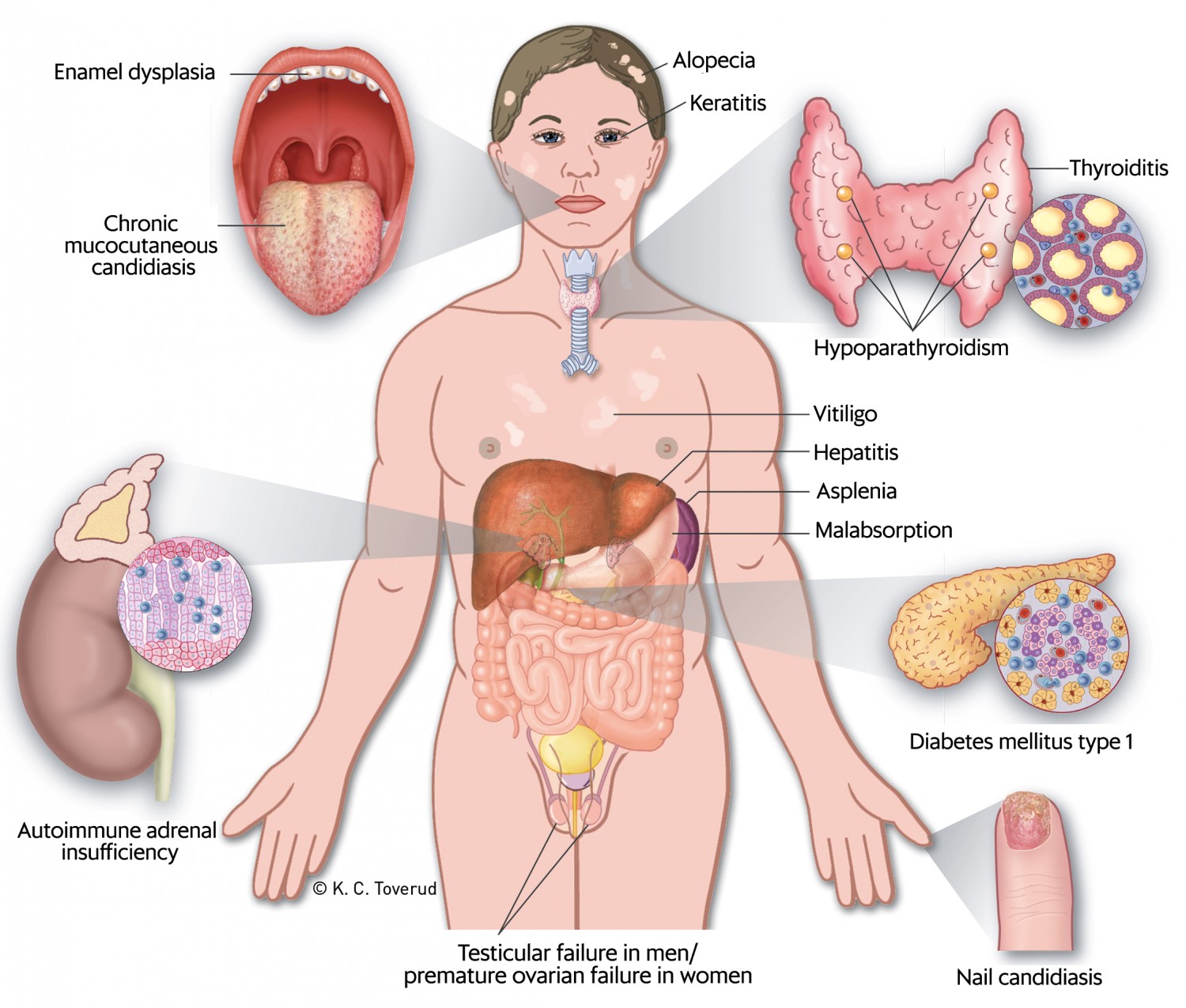
Figure 1 Clinical manifestations that may occur in cases of autoimmune polyendocrine syndrome type 1 (APS-1). The figure has been reproduced with the permission of the authors and the Journal of the Norwegian Medical Association (7).
A diagnosis of autoimmune polyendocrine syndrome type 1 is made if the patient fulfils two of the three criteria of the triad (15, 18). If a sibling has been diagnosed with the syndrome, the diagnosis can be made if one of the criteria is fulfilled (7). Given the variable presentation of symptoms as well as the progressive course, there should be a low threshold for assessing a patient who has one or more of the symptoms in the classic triad. This applies both to paediatricians and to dentists who treat children with recurrent mucocutaneous candidiasis or enamel hypoplasia. The variable presentation can make diagnosis difficult. Because more than 95 % of patients have autoantibodies against interferon (INF)-ω or INF-α (11, 19, 20), measuring these autoantibodies may be a useful screening tool (9). In Norway, this analysis is available at Haukeland University Hospital. A definitive diagnosis is made by sequencing the AIRE gene (9).
There is no curative treatment for autoimmune polyendocrine syndrome type 1. Treatment is symptomatic with replacement of affected hormones and management of complications (9, 16). A number of the manifestations, such as autoimmune hepatitis and bronchiolitis, can be serious in some cases or even fatal. It is also important to treat mucocutaneous candidiasis, as left untreated it increases the risk of squamous cell carcinoma (16).
