Nevrotransmitteren glutamat spiller en viktig patofysiologisk rolle i hjernen. For eksempel vil glutamatakkumulering ekstracellulært under en iskemifase ofte resultere i celledød i det affiserte området. I tillegg synes glutamat å spille en rolle for celletap når det gjelder nevrodegenerative sykdommer som Alzheimers sykdom, Parkinsons sykdom, Huntingtons sykdom og AIDS-demens. Den glutamaterge synapse har derfor vært i sentrum som potensielt terapeutisk mål helt siden den toksiske effekten av glutamat i hjernen ble beskrevet for over 30 år siden (1). Den ble siden gitt betegnelsen eksitotoksisitet.
Den klassiske modellen for eksitotoksisitet sier at overstimulering av glutamatreseptorer (spesielt NMDA-reseptorer) resulterer i en vedvarende intracellulær opphopning av kalsium, som så setter i gang prosesser som ender i celledød. Mulige mekanismer for eksitotoksisitet (2) er beskrevet i figur 1. Rask og massiv intracellulær opphopning av kalsium leder ofte til nekrose, mens andre forstyrrelser i kalsiumomsetningen kan lede til apoptose (3).
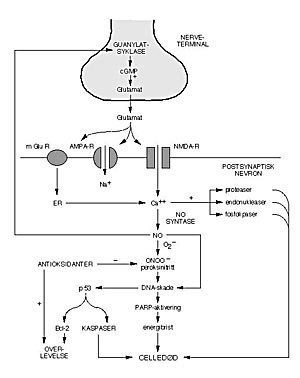
Figur 1 Biokjemiske kaskader aktivert av iskemi (eksitotoksisitetshypotesen). Nervecelledød ved eksitotoksisitet kan forårsakes av overstimulering av glutamatreseptorer. Dette fører til en vedvarende intracellulær kalsiumopphopning og aktivering av kalsiumavhengige prosesser. Pilene viser aktivering av noen mulige mekanismer. + angir aktivering og – angir hemming. AMPA-R = AMPA-subtypen av glutamatreseptorer, cGMP = syklisk GMP, ER = endoplasmatisk retikulum, mGluR = metabotrope glutamatreseptorer, NMDA-R = NMDA-subtypen av glutamatreseptorer, PARP = poly(ADP-ribose)polymerase. Bcl-2 og p53 omhandles i Løberg og medarbeideres artikkel (2)
Prinsipielt kan omfanget av en iskemisk hjerneskade begrenses ved trombolyse eller nevroproteksjon. Til tross for intensiv forskning og kartlegging av molekylære mekanismer for glutamatindusert celledød finnes det ennå ingen etablert behandling som griper inn i signalkaskadene som er initiert av glutamat. For tiden pågår det mer enn 30 kliniske utprøvninger i ulike faser med potensielle nevroprotektive forbindelser, og de fleste av disse er hemmere av eksitotoksisitet (glutamatreseptorantagonister og natrium- og kalsiumkanalblokkere). Mange glutamatreseptorantagonister som virket godt i dyreforsøk har vist skuffende resultater i klinisk utprøvning. Flere av disse er derfor blitt stoppet. En mulig forklaring er at man griper inn for tidlig i signalkaskaden og at man heller burde konsentrere seg om å blokkere signalkaskadene nedstrøms for glutamatreseptorene. Man må også være åpen for at eksitotoksisitet mediert av NMDA-reseptorer kun er én av flere mulige mekanismer som ligger til grunn for iskemisk celledød hos menneske (4).
Kalsiuminnstrømming kan også skje gjennom AMPA-reseptorer (AMPA = α -amino3-hydroksy-5-metyl-4-isoksazolpropionsyre) (4). Andelen AMPA-reseptorer som er permeabel for kalsium- og sinkioner blir større ved iskemi, og det bidrar til å forklare at AMPA-reseptorantagonister gir god beskyttelse av nevroner i visse former for eksperimentell iskemi. Det er nå økende holdepunkter for at sink har en patofysiologisk rolle ved iskemi i hjernen. Redusert nivå av kalium intracellulært er også assosiert med nevronal apoptose.
Nekrose og apoptose synes å bli indusert parallelt i iskemisk hjernevev, og det er mange faktorer som bestemmer hvilken mekanisme som dominerer, bl.a. hvor alvorlig insultet er. Paradoksalt vil et redusert kalsiumnivå intracellulært disponere for apoptotisk celledød. Det er foreslått at eksitotoksisk kalsiumopphopning dominerer akutt i nærheten av det iskemiske kjerneområdet, mens kalsiumdeprivasjon og apoptose dominerer i den iskemiske randsonen på et senere tidspunkt (4). Hvis dette holder stikk, vil en terapi rettet mot kalsiumhomøostasen måtte ha ulik strategi i ulike tidsvinduer. Fremtidens behandling av iskemi vil sannsynligvis bli en stadig mer raffinert akutt trombolytisk behandling, koblet til inhibering av eksitotoksisk nekrose og iskemisk apoptose i ulike tidsvinduer.
Subtypespesifikke glutamatreseptorantagonister kan også bli aktuelt, samt legemidler som påvirker ionehomøostaser (kalsium, kalium og sink) og intracellulære biokjemiske kaskader (hemmere av proteaser, frie oksygenradikaler og induserbare former av NO-syntase). Inngrep i inflammatoriske kaskader vil sannsynligvis også bidra til å redusere celledød. Noen av behandlingsprinsippene, spesielt de som reduserer apoptose, kan også bli aktuelle ved nevrodegenerative sykdommer. For langvarig farmakologisk hemming av apoptose i hjernen vil det være essensielt å utvikle cellespesifikke hemmere for å unngå neoplasi i andre organsystemer.
