Polynevropati (1, 2) skyldes affeksjon av mange perifere nerver. Symptomene kan være sensoriske, motoriske og/eller autonome, og tilstanden kan ha mange årsaker. Vanlige årsaker til polynevropati er diabetes mellitus, høyt alkoholkonsum, autoimmune tilstander, som Guillain-Barrés syndrom, og arvelige former f.eks. Charcot-MarieTooths sykdom. En mindre vanlige årsak til polynevropati er amyloidose.
Amyloidose kan affisere flere organer og skyldes ekstracellulær avleiring av uløselige proteinfibriller (amyloider) bl.a. i perifere nerver (3, 4). Mer enn 15 proteiner kan danne amyloid, med minst 20 forskjellige amyloidosetilstander (3, 4). Amyloidose kan klassifiseres på mange forskjellige måter, men amyloidproteinet brukes ofte som basis (5). De klinisk vanligste amyloidosetilstandene (4) er:
– Reaktiv systemisk amyloidose, med avleiring av AA-amyloid, som forekommer sekundært til kroniske inflammatoriske, infeksiøse eller neoplastiske sykdommer.
– Immunocyttdyskrasiamyloidose, med avleiring av AL-amyloid, forårsaket av B-lymfocyttdyskrasi med eller uten funn av immunglobulin lette kjeder i urin eller blod. Den kan være primær og kalles da primær systemisk amyloidose, eller den kan være assosiert med myelom, lymfom eller makroglobulinemi.
– Senil amyloidose, som oftest er asymptomatisk og finnes i mer eller mindre grad hos alle individer over 80 år.
– Cerebral amyloidose, som finnes f.eks. ved Alzheimers sykdom eller cerebral amyloid angiopati (6).
– Hereditær amyloidose som innbefatter flere familiære former. Familiær amyloidotisk polynevropati (7) er assosiert med autosomalt dominant mutasjon i genet for plasmaproteinet transthyretin (8). En annen hereditær form er den autosomalt recessive sykdom familiær middelhavsfeber (4).
Vi rapporterer her tre tilfeller av primær systemisk amyloidose som debuterte med polynevropati.
Pasienter
Pasient 1. 70 år gammel mann som fire år tidligere ble utredet for lungefortetninger, uten at det var holdepunkter for sarkoidose, tuberkulose eller malignitet. Han var ellers tidligere frisk. Han ble innlagt pga. åtte måneders sykehistorie med tiltakende svie, smerter, parestesier og brenning i begge underekstremitetene, med ustøhet og ortostatisme samt avmagring og vekttap. Han hadde ikke misbrukt alkohol og ikke hatt diabetes mellitus eller vitamin B12-mangel. Klinisk hadde han normale dype senereflekser og vibrasjonssans, hyperestesi/-algesi og allodyni kombinert med moderate pareser i underekstremitetene og betydelig ortostatisk blodtrykksfall. Omfattende utredning viste ikke tegn til malignitet eller annen kronisk infeksiøs eller inflammatorisk sykdom. Elektromyografi (EMG) og elektronevrografi (ENG) viste sensorisk-motorisk polynevropati av blandet demyeliniserende og aksonal type. Det var ikke noen sikre responser å registrere fra de sensoriske nervene i beina, men ledningshastighetene i de motoriske nervene i beina var ca. 37 m/s. Spinalvæskeanalyse viste ingen leukocytter, og spinalprotein var 1,27. Det ble påvist to serumlike IgG-bånd i spinalvæske, samt to bånd i urinen. Han utviklet biventrikulær kardiomegali i løpet av det siste året. Pasienten ble behandlet med høydose steroider og plasmaferese, uten effekt. I nervebiopsi ble det påvist hyalint materiale (fig 1), identifisert som amyloid. Pga. dårlig allmenntilstand ble kjemoterapi ikke instituert.
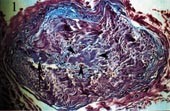
Figur 1 Mikrofotografi av nervefascikkel farget med hematoksylin og eosin. Hyalint materiale påvises i karvegg (pil) og diffust i nervefascikkelen (pilhoder) hos pasient med primær systemisk amyloidose
Pasient 2. 72 år gammel kvinne som tidligere hadde vært frisk bortsett fra strumaoperasjon 35 år tidligere og vitamin B12-substituert kobalaminmangelanemi. Hun hadde ikke misbrukt alkohol og hadde ikke diabetes mellitus. Hun ble innlagt på grunn av progredierende polynevropati som hadde begynt distalt i begge underekstremitetene og migrerte proksimalt, med sensibilitetsutfall, svakhet, parestesier og smerter. Hun hadde også ortostatisme. Symptomene hadde utviklet seg over 2,5 år. Ved klinisk undersøkelse hadde hun normale dype senereflekser og vibrasjonssans, nedsatt sensibilitet for stikk og berøring kombinert med moderate pareser i underekstremitetene samt ortostatisk blodtrykksfall. EMG og ENG ble ikke tatt. Utredningen viste ikke tegn til malignitet, kronisk infeksiøs eller inflammatorisk sykdom. Spinalvæskeanalyse viste 3-4 leukocytter, og spinalprotein var 1,3. Elektroforese av serum, urin og spinalvæske viste ett monoklonalt bånd i gammaregionen. Hun hadde lett forstørret hjerte og tegn til hjerte- og nyresvikt. Hun ble behandlet med høydose steroider, uten effekt. Hun døde plutselig av hjertestans. Obduksjonsrapport viste at pasienten hadde ferskt hjerteinfarkt og i tillegg langt fremskredet primær systemisk amyloidose hovedsakelig med amyloidavleiring i blodkarenes media (fig 2).

Figur 2 Mikrofotografi av blodkar med amyloidavleiring påvist med kongorød farging visualisert med dobbeltbrytning i polarisert lys hos pasient med primær systemisk amyloidose
Pasient 3. 57 år gammel mann som tidligere stort sett hadde vært frisk. Han ble innlagt pga. ni måneders sykehistorie med tiltakende smerter og parestesier i begge underekstremitetene, spesielt i anklene og fotsålene, med ustøhet og ortostatisme samt 20 kg vekttap. Han hadde ikke misbrukt alkohol og ikke hatt diabetes mellitus eller vitamin B12-mangel. Klinisk hadde han utslukkede akillesreflekser, men ellers normale dype senereflekser og vibrasjonssans, nedsatt sensibilitet for stikk og berøring i underekstremitetene samt parese for dorsalfleksjon i anklene. Han hadde også betydelig ortostatisk blodtrykksfall. Omfattende utredning viste ikke tegn til malignitet, eller kronisk infeksiøs eller inflammatorisk sykdom. EMG og ENG viste sensorisk-motorisk polynevropati av aksonal type, og det var ikke noen responser å registrere fra sensoriske eller motoriske nerver i beina. Spinalvæskeanalyse viste ingen leukocytter, og spinalprotein var 1,9. Serum-elektroforese var normal, men i urinen ble det påvist svakt monoklonalt bånd, identifisert som lette kjeder type lambda. I nervebiopsi ble det påvist amyloidose og spesifikk immunfarging påviste lette kjeder type lambda (fig 3). Pasienten ble behandlet med prednisolon og melfalan. Senere utviklet han nyresvikt, og 2,5 år etter debut av polynevropati døde han av hjertestans etter kortvarig synkope.
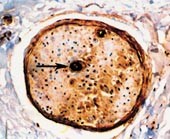
Figur 3 Mikrofotografi av nervefascikkel med immunopositivitet for lambda lette kjeder hos pasient med primær systemisk amyloidose. Den kraftigste fargingen (pil) sees i blodkar
Diskusjon
De tre pasientene hadde et likeartet sykdomsforløp med smertefull, sensorisk-motorisk polynevropati som rammet begge underekstremitetene, samt at det forelå autonom affeksjon. Pasient 1 og pasient 2 hadde lett hjerteforstørrelse og tendens til hjertesvikt. Pasient 2 og pasient 3 fikk i tillegg nyresvikt. Monoklonalt bånd i serum og/eller urin ble funnet hos pasient 2 og 3, mens det var oligoklonale bånd hos pasient 1. Monoklonale bånd forekommer i serum og/ eller urin hos 88 % av pasientene med primær systemisk amyloidose (9). EMG og ENG ble ikke utført hos pasient 2, men viste sensorisk-motorisk polynevropati av aksonal type hos pasient 3, og blandet demyeliniserende og aksonal type hos pasient 1. Ikke hos noen av pasientene var det tegn til arvelig sykdom, malignitet, kronisk infeksiøs eller inflammatorisk sykdom. Amyloidose ble bekreftet histologisk hos de tre pasientene. I alle tilfellene var diagnosen primær systemisk amyloidose med polynevropati som første symptom.
Ved systemisk amyloidose (primær eller myelomassosiert) er polynevropati et ikke uvanlig funn, det forekommer i ca. 20 % av tilfellene (9, 10), ofte før diagnosen primær systemisk amyloidose blir stilt (11).
I en studie publisert i 1998 (12) var de vanligste nevrologiske debutsymptomene som ledet til diagnosen primær systemisk amyloidose følgende: parestesier (81 %), autonome symptomer (65 %) og muskelsvakhet (65 %). Diagnosen amyloidose ble stilt sent i sykdomsforløpet, gjennomsnittlig 29 måneder etter debut av polynevropati (12). Hos våre pasienter var også sensoriske og autonome symptomer de mest vanlige. Diagnosen primær systemisk amyloidose ble stilt retrospektivt hos pasient 2, mens det tok ca. år fra debutsymptomene til amyloidose ble påvist hos pasient 1 og 3.
Polynevropati ved primær systemisk amyloidose skiller seg fra nevropati ved andre monoklonale gammopatitilstander inklusive MGUS (monoklonal gammopati med uavklart signifikans), POEMS (polynevropati-organomegali-endokrinopati-monoklonal gammopati syndrom), myelom og Waldenströms sykdom (13 – 15). Ved primær systemisk amyloidose er polynevropatien progredierende og smertefull med autonomt engasjement, og EMG og ENG viser vanligvis primær aksonal degenerasjon, som hos pasient 3. Dette er typiske funn ved tynnfibernevropati (2).Ved andre monoklonale gammopatier med polynevropati er forløpet som regel mer stabilt, uten store smerter og uten autonome symptomer, og nevrofysiologisk undersøkelse viser oftest primær segmentell demyelinisering.
Det finnes ingen effektiv behandling av amyloidose. Gjennomsnittlig overlevelsestid uten behandling er ca. 18 måneder etter diagnosetidspunkt, men med kjemoterapi (melfalan og prednisolon) kan overlevelsen forlenges til ca. 38 måneder (12). Pasient 3 fikk kjemoterapi, men levde bare 18 måneder etter at amyloidose ble påvist. Pasient 2 fikk ingen spesifikk behandling. Begge pasientene døde av hjertestans som følge av hjerteaffeksjon/autonom affeksjon. Symptomatisk behandling av smerter og autonom dysfunksjon må forsøkes, men er ikke alltid effektivt (12).
Primær systemisk amyloidose må vurderes som differensialdiagnose ved progredierende smertefull polynevropati med autonom affeksjon. Diagnosen kan lett stilles ved hjelp av biopsi av rektalslimhinne, fett, beinmarg eller perifer nerve (oftest n. suralis) for undersøkelse av amyloidavleiring. Nevrologiske manifestasjoner er også meget vanlige ved hereditær amyloidose, som bør utelukkes etter amyloidpåvisning i biopsi.
