Artikkelen er tidligere publisert i Ugeskrift for Læger 2000; 162: 1525 – 7
Porfyrisygdommene er metabolisk og biokemisk relateret til biosyntesen af hæm, der ud over at indgå i hæmoglobin og myoglobin også indgår i cytokromerne og i enzymerne peroxidase og katalase. Hæm er udviklingsmæssigt et gammelt molekyle, som findes i alle eukaryote og i de fleste prokaryote celler. Syntesen af hæm starter og slutter i mitokondrierne. Hvert af de otte trin i syntesen er katalyseret af et specifikt enzym. Nedsat aktivitet af et af disse kan medføre en porfyrisygdom (fig 1).
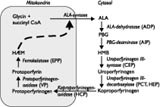
Figur 1 Hæmsyntesevejen. De involverede enzymer er angivet med kursiv og de relaterede porfyrisygdomme med forkortelse i parentes. ALA: 5-aminolævulinat, HMB: Hydroxy methyl bilan, PBG: Porfobilinogen, ADP: Aminolævulinat dehydratase-porfyri, AIP: Akut intermitterende porfyri, CEP: Kongenit erytropoietisk porfyri, PCT: Porphyria cutanea tarda (og den homozygote form, HEP: Hepato-erytropoietisk porfyri), HCP: Hereditær koproporfyri, VP: Variegat porfyri, EPP: Erytropoietisk protoporfyri
Klassifikation af porfyrisygdomme
Porfyrisygdommene har siden århundredets begyndelse været genstand for forskellige – ofte indbyrdes modstridende – klassifikationsforsøg, som overvejende har været baseret på kliniske forløbsformer kombineret med påvisning af forskellige metabolitter i urin og fæces. Den nugældende klassifikation (1) er baseret på aktuel enzymdefekt, organlokalisation og klinisk forløbsform og fremgår af tabell 1. Opdelingen i akutte og ikke-akutte porfyrier refererer til forekomsten af episoder med bl.a. abdominalsmerter, perifer neuropati og mental dysfunktion ved de akutte former (skyldes bl.a. ophobning af 5-aminolævulinat [ALA], der er en strukturel analog til neurotransmitteren 4-aminobutyrat [GABA]). Ved både akutte og ikke-akutte porfyrier kan der forekomme fototoksiske hudmanifestationer, der skyldes ophobning af porfyrinderivater i huden. I praksis omfatter de akutte porfyrier sygdommene akut intermitterende porfyri (AIP), hereditær koproporfyri (HCP) og variegat porfyri (VP), idet de øvrige er ekstremt sjældent forekommende.
Tabell 1 Porfyrisygdommenes inddeling, arvegang, kromosomale placering, genstruktur og hovedklasser af symptomer
|
Symptomer
|
|
Porfyrisygdom
|
Afficeret enzym
|
Arvegang
|
Genlokalisation
|
Genstruktur
|
neurologiske
|
kutane
|
|
Akutte, hepatiske
|
|
Aminolævulinat dehydratase-porfyri
|
5-ALA dehydratase
|
AR
|
9q34
|
13 kb, 13 exons
|
+
|
+
|
|
Akut intermitterende porfyri
|
PBG-deaminase
|
AD
|
11q24.1-24.2
|
10 kb, 15 exons
|
+
|
|
|
Hereditær koproporfyri
|
Koproporfyrinogenoxidase
|
AD
|
3q12
|
14 kb, 7 exons
|
+
|
(+)
|
|
Variegat porfyri
|
Protoporfyrinogenoxidase
|
AD
|
1q21-23
|
4,7 kb, 13 exons
|
+
|
(+)
|
|
Ikke akutte, hepatiske
|
|
Porphyria cutanea tarda
|
Uroporfyrinogen III-decarboxylase
|
KomPleks
|
1p34
|
3 kb, 10 exons
|
|
+
|
|
Ikke akutte, erytropoietiske
|
|
Erytropoietisk protoporfyri
|
Ferrokelatase
|
AD
|
18q21.3
|
45 kb, 11 exons
|
|
+
|
|
Kongenit erytropoietisk porfyri
|
Uroporfyrinogen III-syntase
|
AR
|
10q25.2-26.3
|
45 kb, 10 exons
|
|
+
|
Hyppighed
AIP anses for at være den hyppigst forekommende akutte porfyrisygdom her i landet. Antallet af anlægsbærere i Danmark kendes ikke, men estimater af hyppigheden i Finland (2) og Frankrig (3) baseret på undersøgelser af PBG-deaminaseaktivitet i erytrocytlysater fra bloddonorer tyder på, at frekvensen er mindst 1 : 2.000. Kun 10 – 20 % heraf vil nogensinde få kliniske symptomer. Hyppigheden af HCP og VP i Danmark kendes ikke.
Symptomer
Ved langt de fleste akutte porfyrianfald ses voldsomme abdominalsmerter, ofte ledsaget af opkastninger og obstipation (4). I mere end halvdelen af tilfældene forekommer neuropsykiatriske symptomer som kraftnedsættelse og smerter i ekstremitetsmuskulaturen, pareser og evt. psykoser. I ca. 15 % af tilfældene er anfaldene ledsaget af epileptiske anfald og paræstesier. I en del tilfælde ses takykardi og/eller arteriel hypertension. Af og til ses moderat temperaturforhøjelse og leukocytose. De fleste porfyrianfald remitterer spontant i løbet af nogle dage, men et mindre antal patienter udvikler i relation til anfaldet irreversible neurologiske udfaldssymptomer svarende til en akut polyradiculitis.
Gennem de senere år er der publiceret en række tilfælde af primær hepatocellulær cancer hos patienter med akut porfyri. Således synes helt op til 25 % af patienterne med AIP i Nordsverige at udvikle denne sjældne type af cancer (5). Dette har i Sverige fundamentalt ændret håndteringen af disse patienter, som nu følges regelmæssigt med et kontrolprogram, som har til formål at forebygge porfyriattakker og at afsløre komplikationer på et tidligt tidspunkt, hvor muligheden for intervention er optimal (5). I en ny dansk/svensk registerundersøgelse bekræftes denne overhyppighed af hepatocellulært karcinom hos patienter med AIP og hos patienter med den ikke akutte porfyriform, porphyria cutanea tarda (6). Også hos patienter med hereditær koproporfyri (7) og variegat porfyri (8) er der beskrevet tilfælde af primært hepatocellulært karcinom. En mulig forklaring på den øgede forekomst af hepatocellulært karcinom hos patienter med akut porfyri er den øgede akkumulering af porfyrinmetabolitter, som kan forårsage en endogen produktion af karcinogene substanser via autooxidering af ALA (9).
Genetiske faktorer
Porfyrisygdommene arves autosomalt dominant (bortset fra de to yderst sjældne, recessivt arvelige porfyriformer aminolævulinat dehydratase-porfyri og kongenit erytropoietisk porfyri, som ikke omtales yderligere i denne artikel). Hvis der er mutationer i den ene allel på locus for et af hæmsyntesevejens enzymer, er resultatet ofte, at genproduktet fra denne allel ikke har fuld aktivitet. Dette disponerer personen for porfyrisygdom, men selv mutationer, som medfører en stærkt nedsat aktivitet af et af disse enzymer behøver ikke nødvendigvis at medføre symptomer.
Generne for alle de i hæmsyntesen involverede enzymer er klonede gennem de sidste ti år (Tabel 1). I enkelte områder af verden har man befolkningsgrupper med fælles genetisk ophav, hvor porfyri næsten altid er forårsaget af den samme mutation (eng. founder effect ). Dette kendes ved akut intermitterende porfyri i befolkningen i Nordsverige (10) og ved variegat porfyri hos sydafrikanere (11). De fleste tilfælde af de enkelte porfyrisygdomme forårsages imidlertid af en lang række forskellige mutationer, således at hver familie i princippet har hver sin mutation, såkaldt ”private” mutationer.
Udløsende faktorer
Hos den disponerede patient kan et porfyrianfald udløses af en række forskellige faktorer som faste, stress, endogene hormonsvingninger, alkohol i større mængder og visse lægemidler. De involverede mekanismer er ikke endeligt afklaret, men et fælles træk synes at være en induktion af cytokrom P450 med deraf følgende øget behov for hæm. En øget hæmsyntese vil, såfremt enzymaktiviteten i et af trinene er reduceret, bevirke en ophobning af en eller flere metabolitter. Ved arvelig disposition for akut intermitterende porfyri vil der således pga. den nedsatte porfobilinogen (PBG)-deaminaseaktivitet forekomme en ophobning af PBG og ALA. Ved arvelig disposition for koproporfyri og variegat porfyri vil der på samme måde kunne forekomme en ophobning af koproporfyrinogen og protoporfyrinogen, som begge nedsætter aktiviteten af PBG-de aminase med deraf følgende sekundær ophobning af PBG og ALA. Mange forskellige lægemidler er rapporteret at virke porfyrinogent. For en oversigt over, hvilke lægemidler, der menes at kunne fremkalde porfyrianfald, og hvilke lægemidler der erfaringsmæssigt ikke gør det, henvises til den seneste liste (12). En tilsvarende liste ajourføres af ”Det Danske Porfyricenter” og forventes tilgængelig via Internettet fra foråret 2000.
Diagnostik
Ved akutte anfald
Diagnosen akut porfyri (AIP, HCP, VP) er baseret på familieanamnese, kliniske symptomer og påvisning af forøget udskillelse af ALA og PBG i urinen. ALA og PBG bør altid bestemmes kvantitativt, idet kvalitativ påvisning af PBG i urinen ved hjælp af Ehrlichs aldehydreagens kun er positiv, såfremt koncentrationen af PBG er større end ca. 100 µ mol/l (referenceværdier: ALA< 40 µ mol/døgn, PBG< 15 µ mol/ døgn). Normal udskillelse af ALA og PBG i urinen udelukker porfyrisygdom som årsag til patientens symptomer. Da behandlingen af et akut porfyrianfald er uafhængig af, om det drejer sig om AIP, HCP eller VP, kan denne baseres alene på kvalitativ bestemmelse af U-ALA og U-PBG. Efterfølgende suppleres med bestemmelse af porfyrinudskillelsesmønsteret i urin og – ved mistanke om HCP eller VP – i fæces. Nedsat aktivitet af PBG-deaminase i erytrocytter understøtter diagnosen AIP.
Uden for anfald
AIP: Mindre end 20 % af personerne med hereditær disposition for AIP har uden for anfald forhøjet udskillelse af ALA og PBG. Under forudsætning af normal erytrocytaldersfordeling, udelukker normal aktivitet af PBG-deaminase i erytrocytterne, medmindre det drejer sig om en af de meget sjældne mutationer i exon 1, hereditær disposition for AIP. Såfremt familiens mutation er kendt, baseres undersøgelsen for arvelig disposition for AIP på molekylærgenetiske principper.
HCP: Antallet af personer med hereditær disposition for HCP, der uden for anfald har forøget koproporfyrinudskillelse i urin og/eller fæces, kendes ikke. Såfremt familiens mutation er kendt, baseres undersøgelsen for arvelig disposition for HCP på molekylærgenetiske principper.
VP: Kun få personer med hereditær disposition for VP har uden for anfald forøget udskillelse af protoporfyrin i fæces. Såfremt familiens mutation er kendt, baseres undersøgelsen for arvelig disposition for VP på molekylærgenetiske principper.
Molekylærgenetisk udredning
Kendskab til sygdommenes genetiske grundlag har afgørende betydning for valg af metode til den molekylærgenetiske diagnostik. Såfremt familiens mutation ikke er kendt, er det vigtigt, at de molekylærgenetiske undersøgelser udføres på en proband med så klare og entydige symptomer/biokemiske fund som muligt. Sygdommen udviser stor allelheterogenitet, og som i andre undersøgte befolkningerne har vi i den danske befolkning kun i få tilfælde fundet ubeslægtede AIP-familier med samme mutation i PBG-deaminasegenet. Det er derfor nødvendigt a priori at gå ud fra, at en ny familie har sin egen ”private” mutation i det relevante gen.
Påvisning af ”private” mutationer kræver i princippet afsøgning af hele genet, da man ikke på forhånd kan udpege et område af genet, som med særlig sandsynlighed indeholder en sygdomsrelateret mutation. Til afsøgning af hele genområdet for mutationer er der i det væsentlige to fremgangsmåder: Enten direkte sekventering af genet eller påvisning af allelvariationer med anvendelse af en ”screeningsteknik” efterfulgt af sekventering af et eller nogle få områder af genet udpeget med ”screeningsteknikken”. Ved anvendelse af denne strategi reduceres sekventeringsarbejdet meget væsentligt, hvilket er af stor betydning, da sekventering udgør et nåleøje for de fleste laboratorier. Ved Det Danske Porfyricenter foretages ”genscreening” med denaturerende gradient gel-elektroforese (DGGE)-teknikken (13) til mutationsundersøgelse af DNA fra patienter, hos hvem man mistænker AIP, hereditær koproporfyri, porphyria cutanea tarda og variegat porfyri.
Behandling og monitorering
Generelt anvendes kun medikamenter, der vides ikke at kunne fremkalde porfyrianfald. For at supprimere glukoneogenesen gives under anfald isoton glukoseopløsning i.v. (3 – 4 l/døgn). Mod smerter gives pethidin eller morphfin. Ved sværere anfald kan anvendes hæmarginat, der via negativ feedback hæmmer hæmsyntesens første trin. Effekten af den givne behandling kan evt. følges ved måling af indholdet af ALA og især PBG i urinen. En del patienter kan også selv lære at håndtere mindre porfyrianfald ved peroral indtagelse af glukoseholdige væsker.
På grund af den forøgede forekomst af hepatocellulært karcinom hos disse patienter anbefaler nogle (5) årlig UL-scanning af leveren og eventuelt måling af alfaføtoprotein i serum fra 55-års-alderen.
Håndtering af porfyrisygdomme i Danmark
De akutte porfyrisygdomme hører til blandt de sjældne metaboliske sygdomme. Som en konsekvens heraf blev ”Det Danske Porfyricenter” etableret omkring 1990 med det formål at samle den biokemiske og molekylærbiologiske diagnostik og herved sikre tilstrækkelig kompetence og erfaringsgrundlag.
Centeret har siden da udviklet sig og omfatter i dag klinisk biokemisk afdeling, Viborg Sygehus (metabolitter og enzymer) og en række afdelinger på Odense Universitetshospital: Afdeling KKA, klinisk biokemi (molekylærbiologisk diagnostik) og klinisk genetik (genetisk rådgivning), endokrinologisk afdeling M (diagnostik, behandling og rådgivning vedrørende medikamenter) og dermatologisk afdeling I (diagnostik og behandling af kutane porfyrier).
I DNA fra 31 familier henvist til centeret på mistanke om akut porfyrisygdom fandtes hos 60 personer mutation i PBG-deaminasegenet (15 forskellige mutationer), mens tre havde mutation i koproporfyrinogen-oxidasegenet (to forskellige mutationer). Mange af de karakteriserede mutationer er kun eller først påvist i DNA fra de danske probander. Ud over diagnostik og behandling af akutte og ikke-akutte porfyrisygdomme kan centeret tilbyde kontakt til andre specialkyndige kolleger.
Centerets forskning, som foregår i samarbejde med forskere i udlandet, omfatter bl.a. udvikling af nye biokemiske og molekylærgenetiske metoder til diagnostik af de forskellige porfyrisygdomme, studier af genotype-fænotyperelationer på enzymniveau, cellulært niveau og i relation til det kliniske forløb samt studier af porfyrisygdommenes patogenetiske forhold.
Det Danske Porfyricenter, som foruden de to anførte klinisk biokemiske afdelinger, omfatter følgende afdelinger på Odense Universitetshospital: Afdeling KKA, klinisk genetik, endokrinologisk afdeling M og dermatologisk afdeling I.
