Primær immunsvikt-sykdommer (PID) representerer en gruppe oftest medfødte sykdommer som i de fleste tilfeller skyldes mutasjoner i gener av essensiell betydning for normal utvikling og/eller funksjon av viktige celler i immunsystemet. Tabell 1 gir en forenklet oversikt over de viktigste hovedgrupper av primær immunsvikt-sykdommer som er kjent i dag (1). De fleste sykdommer i denne gruppen er sjeldne. I løpet av den siste tiårsperioden er den genetiske defekt påvist for en rekke primær immunsvikt-sykdommer, og de aktuelle geners funksjon i immunsystemet er også i mange tilfeller kartlagt. Det dreier seg ofte om meget alvorlige sykdommer, hvor kurativ behandling i de fleste tilfeller ikke finnes, bortsett fra i de tilfeller hvor beinmargstransplantasjon er mulig. Primær immunsvikt-sykdommer ansees derfor som et attraktivt mål for genterapi, og det var ved en av disse sykdommene genterapi første gang ble forsøkt klinisk i 1990.
|
Tabell 1 Primær immunsvikt-sykdommer. De viktigste hovedgrupper
|
|
|
Inkluderer blant annet
|
|
I
|
Kombinert B- og T-cellesvikt
|
Forskjellige varianter av SCID (alvorlig kombinert immundefekt) Hyper IgM-syndrom
|
|
II
|
Defekter som vesentlig rammer antistoffproduksjonen
|
Kjønnsbundet recessiv agammaglobulinemi (Bruton-type) Vanlig variabel immunsvikt 1 Selektive IgG-subklassedefekter Selektiv IgA-mangel
|
|
III
|
Defekter i fagocyttfunksjonen
|
Kronisk granulomatøs sykdom Kongenitale nøytropenier Leukocyttadhesjonsdefekter
|
|
IV
|
Defekter i komplementsystemet
|
Defekter i komplementkaskadens proteiner Defekter i komplementregulerende proteiner (f.eks. hereditært angioødem)
|
|
V
|
Defekter i lymfocyttapoptose
|
Autoimmunt lymfoproliferativt syndrom
|
|
VI
|
Syndromer assosiert med DNA- brudd
|
Ataxia teleangiectasia hereditaria
|
|
VII
|
Interferon γ -assosierte immunsviktsykdommer
|
Interferon γ - og IL-12-reseptordefekter
|
|
VIII
|
Andre veldefinerte immunsviktsyndromer
|
Wiskott-Aldrichs syndrom Autoimmun polyendokrinopati med candidiasis og ektodermal dystrofi DeGeorges syndrom Duncans syndrom (defekt i responsen på EBV-antigener) Jobs syndrom
|
|
|
I dag er kliniske forsøk med genterapi i gang ved flere primær immunsvikt-sykdommer, og det foreligger preliminære kliniske resultater fra flere av disse forsøkene. Hittil er ingen primær immunsviktsykdom helbredet med genterapi, men erfaringene fra utførte undersøkelser har gitt verdifull innsikt for videre arbeid på dette feltet. Vanskeligheter som må overvinnes, er dels knyttet til generelle problemer ved human genterapi, dels til spesielle forhold knyttet til immunsystemets kompleksitet.
Forutsetning for utvikling av genterapi
Før man kan satse på utvikling av genterapi for en bestemt primær immunsvikt-sykdom, er det en rekke betingelser som må oppfylles (2, 3). For det første bør genet være nøye studert, slik at DNA-sekvensen og organiseringen av genomet er klarlagt. For flere immunsviktsykdommer, for eksempel ”common variable immunodeficiency”, som er den hyppigste årsak til primær hypogammaglobulinemi, er disse betingelsene ennå ikke oppfylt. Videre bør genets funksjon være kjent, da manipulasjon av genet ellers ville kunne ha uante konsekvenser. Betydelige problemer er også knyttet til de spesielle reguleringsmekanismer som er operative for mange essensielle gener i immunsystemet.
Noen gener som ville være attraktive å angripe ved genterapi, uttrykkes bare i spesielle celletyper, for eksempel det såkalte BTK-genet i B-lymfocytter, eller bare i en spesiell funksjonell eller utviklingsmessig fase av en celletype, som for eksempel CD40L-genet i aktiverte T-lymfocytter. Genterapiteknologien i dag tillater ikke en slik selektiv regulering av genets uttrykk in vivo, og genterapi vil derfor kunne få alvorlige konsekvenser hvis det terapeutiske gen og genprodukt uttrykkes i de ”gale” celler på ”gale” tidspunkter under en immunrespons. I en dyreeksperimentell modell er det f.eks. vist at økt ekspresjon av CD40L kan forårsake lymfoproliferativ sykdom.
For visse geners vedkommende er det også en stram fysiologisk regulering av genekspresjonen kvantitativt, og i slike tilfeller vil en genterapeutisk manipulering som fører til vesentlig høyere ekspresjon av genet og dets genprodukt enn normalt, også kunne medføre alvorlige bivirkninger. Omfattende cellekultureksperimenter er utført med mange av de gener som er identifisert ved primær immunsvikt-sykdommer. Slike eksperimenter må, så sant det er mulig, suppleres med studier i egnede dyremodeller, som nå finnes for flere viktige primær immunsvikt-sykdommer, før kliniske forsøk med genterapi startes ved de enkelte immunsviktsykdommer.
Aktuelle strategier for genterapi
I de kliniske forsøk som er i gang ved genterapi av primær immunsvikt-sykdommer, er det to celletyper som er blitt definert som målceller for genoverføring: hematopoetiske stamceller og lymfocytter .
Siden førstnevnte er opphavscellen for alle de øvrige celler i immunsystemet og fornyer seg selv, ville det i utgangspunktet synes mest hensiktsmessig å foreta genmanipulering av denne celletypen. Genoverføringen til disse cellene er imidlertid lite effektiv med de eksisterende vektorsystemer, og dette har hemmet utviklingen på feltet.
Siden lymfocytter er essensielle effektorceller ved immunologiske reaksjoner, og defekten ved primær immunsvikt-sykdommer ofte sitter i lymfocyttene, er denne celletypen også en naturlig målcelle ved genterapi. Et problem her er imidlertid at antall tilgjengelige lymfocytter er sterkt nedsatt ved flere av de alvorligste former for primær immunsvikt-sykdom, og dette begrenser mulighetene for å anvende lymfocytter som målceller. Ved visse former for primær immunsvikt-sykdom sitter defekten i monocytter , som derfor også er en potensiell målcelle for genterapi. Et alvorlig problem her er den korte levetid slike celler har, uten at de fornyer seg ved celledeling og gir det terapeutiske genet videre. De eksisterende kliniske erfaringer er alle gjort med genterapi rettet mot lymfocytter eller CD34-positive hematopoetiske stamceller.
Nedenfor gis en kort omtale av de formene for primær immunsviktsykdom hvor kliniske forsøk med genterapi allerede har vært forsøkt eller er i gang.
Alvorlig kombinert immundefekt (severe combined immunodeficiency, SCID)
SCID er samlebetegnelsen på en gruppe alvorlige primær immunsvikt-sykdommer som er assosiert med funksjonssvikt av både B- og T-cellesystemet i immunapparatet, av og til også av de såkalte cytotoksiske NK-cellene. Dette fører til en svikt i hele det immunologiske infeksjonsforsvar som manifesterer seg allerede i de første seks måneder etter fødselen, med alvorlige infeksjoner med bakterier, sopp (særlig Candida, Pneumocystis carinii) og virus. Pasienten har ofte gastrointestinale symptomer med vedvarende diaré og generelt dårlig trivsel og utvikling.
I hvert fall seks forskjellige genmutasjoner er nå beskrevet som årsak til SCID. De har forskjellig arvegang og enkelte ulikheter i immunologisk defektmønster, mens de kliniske konsekvenser ofte er nokså like. Felles for alle SCID-variantene er at de er dødelige uten terapi, som er beinmargstransplantasjon ved alle former, unntatt ved SCID som skyldes defekt i enzymet adenosindeaminase (ADA). Kun ved ADA-defekt foreligger et alternativ til beinmargstransplantasjon, nemlig substitusjonsbehandling med ADA koblet til polyetylenglykol (PEG-ADA), som må gis parenteralt tre ganger per uke. Dette er en meget kostbar behandling som ikke er kurativ.
Adenosindeaminasemangel
Mangelen på enzymet adenosindeaminase (ADA) er en viktig årsak til autosomal recessiv SCID. Allerede i midten av 1980-årene ble denne tilstanden identifisert som en svært velegnet sykdom for genterapi, og er den første sykdom hos menneske hvor slik terapi er forsøkt klinisk (4). Fordelen ved denne sykdommen som kandidat for genterapi er blant annet at ADA-genet er grundig undersøkt, og dets funksjon og regulering er kjent. Årsaken til SCID ved ADA-mangel er at enzymdefekten fører til progredierende akkumulering av den toksiske purinmetabolitten deoksyadenosin i lymfocyttene, som fører til mangel på B- og T-celler, mens det av og til kan påvises funksjonelle NK-celler.
En fordel ved genterapeutiske forsøk ved ADA-mangel er at humane T-lymfocytter fungerer normalt over et vidt spektrum av ulike ADA-konsentrasjoner (fra 5 % av det normale til omkring 50 ganger den normale gjennomsnittskonsentrasjonen), og bivirkninger av for høy genekspresjon skulle derfor ikke være noe problem. Etter vellykkede prekliniske studier med cellekulturer ble kliniske forsøk med genterapi startet i 1990 og foregår nå etter fem ulike behandlingsprotokoller (2, 3) Støttebehandling med PEG-ADA har vært gitt til pasientene. I to av behandlingsstudiene, inkludert den første, er T-lymfocytter fra perifert blod blitt benyttet som målceller. I en av de øvrige har hematopoetiske stamceller fra beinmarg vært benyttet, i en annen CD34-positive stamceller isolert fra navlestrengsblod og i den siste både hematopoetiske stamceller og perifere T-lymfocytter.
Den foreløpige konklusjonen fra behandling av rundt ti pasienter er at ingen alvorlige bivirkninger har vært knyttet til de genterapeutiske prosedyrene. Ikke hos noen av pasientene synes helbredelse å ha inntrådt, og man har heller ikke kunnet avslutte substitusjonsbehandlingen med PEG-ADA. Dette kompliserer evalueringen av behandlingseffekten. Forsøkene har imidlertid vist at genetisk modifiserte T-lymfocytter i flere tilfeller kan påvises i organismen i flere år etter den genterapeutiske prosedyre (5). Observasjoner tyder også på at de cellene som har fått overført det normale ADA-genet, har en overlevelsesfordel i organismen sammenliknet med tilsvarende celler med defekte ADA-gener, og på lengre sikt vil da forhåpentlig den normale celletypen øke i andel (6). Bare videre observasjoner med forsøk på seponering av PEG-ADA-behandlingen vil gi svar på om genterapi etter de foreliggende protokoller har vesentlig klinisk verdi ved denne formen av SCID.
Kjønnsbundet recessiv SCID
Ved denne formen av SCID foreligger en mutasjon i γ -kjeden som inngår i lymfocyttreseptoren for interleukin 2. Det viser seg at denne γ -kjeden også er en komponent i lymfocyttreseptorene for flere andre viktige cytokiner, blant annet IL-4, IL-7, IL-9 og IL-15 (fig 1). Nye studier tyder på at denne felles γ -kjeden ( γ c) er vesentlig for intracellulær videreføring av aktiveringssignalet etter stimulering av den respektive cytokinreseptor på overflaten. Mutasjoner i γ c-kjeden fører derfor til fundamentale forstyrrelser i lymfocyttenes respons på en rekke viktige cytokiner som også er essensielle i T- og B-cellers normale vekst og differensiering. Disse pasientene har sterkt nedsatt antall T-celler, men normalt eller sågar økt antall B-lymfocytter, som imidlertid ikke viser normal funksjon. Den eneste kurative behandling som finnes, er beinmargstransplantasjon, som kan gi meget gode resultater, særlig med en fullt forlikelig giver. Ikke sjelden
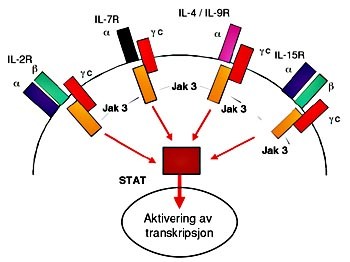
Figur 1 Figuren viser oppbyggingen av membranreseptorene for cytokinene IL-2 (IL-2R), IL-7 (IL-7R), fellesreseptoren for IL-4 (IL-4/IL-9R) og IL-9, samt reseptoren for IL-15 (IL-15R). Samtlige reseptorer består av to eller tre polypeptidkjeder, hvorav én kjede, γ c (gamma common), er felles for alle. Når reseptorene aktiveres via binding av det korresponderende cytokin, skjer signaloverføringen ved at reseptorsignalet overføres fra γ c til Janus-kinase 3 (Jak 3), som aktiverer STAT-proteiner (STAT = signal transducer and activator of transcription). STAT-proteinene vandrer så til cellekjernen og aktiverer transkripsjonsprosesser som er essensielle for normal lymfocyttfunksjon. Mutasjoner i genet for γ c setter alle de nevnte cytokinreseptorer ut av spill og fører til alvorlig immundefektsyndrom (SCID). Av figuren fremgår også at mutasjoner i genet for Jak 3 kan føre til SCID. Også mutasjoner i α -kjeden i IL-7R er beskrevet som årsak til SCID
vil imidlertid resultatet av beinmargstransplantasjon være bare ufullstendig immunologisk normalisering, og utvikling av genterapi har derfor vært ansett som svært ønskelig.
Prekliniske forsøk i cellekulturer har vist at overføring av det normale gen til hematopoetiske stamceller er mulig med godt funksjonelt resultat (2). Et visst gjennombrudd skjedde for kort tid siden, da det ble rapportert gunstige resultater av genterapi med en retroviral vektor assosiert med det normale γ c-genet hos to spedbarn på henholdsvis 8 og 11 måneder (7). Barna synes etter riktignok relativt kort tids oppfølging (ti og 11 måneder) å utvikle seg normalt uten sykdomstegn og viser tegn på betydelig immunologisk normalisering, spesielt når det gjelder T- og NK-lymfocytter.
SCID forårsaket av defekt i genet for JANUS-kinase 3 (JAK 3)
En annen variant av autosomal recessiv SCID skyldes en mutasjon i det viktige intracellulære enzymet Janus-kinase 3 (JAK 3). Dette enzymet har en sentral funksjon ved intracellulære signaloverføringer etter stimulering av overflate reseptorene for cytokinene i IL-2, IL-4, IL-7, IL-9 og IL-15, som skjer via disse reseptorenes felles γ c-kjede, som beskrevet ovenfor (fig 1). Defekter i JAK-3-enzymet gir en autosomalt recessiv SCID, som, hva manifestasjoner angår, er nærmest identisk med kjønnsbundet recessivt SCID som beskrevet ovenfor. Også ved JAK 3-defekt SCID er beinmargstransplantasjon den eneste livreddende behandling, men er ikke alltid mulig. Eksperimenter i cellekulturer har vist at det lar seg gjøre å overføre det normale gen til hematopoetiske stamceller, og ganske nylig er det vist at JAK 3-genet kan overføres til stamceller fra knockoutmus med JAK 3-defekt, og at tilbakeføringen av slike stamceller fører til betydelig immunologisk normalisering (2). Også ved denne tilstanden er en klinisk protokoll godkjent og behandlingsforsøk er i gang. Ingen resultater foreligger ennå.
Mangel på purinnukleosidfosforylase
En annen sjelden årsak til livstruende primær immunsvikt-sykdom er en genetisk defekt i enzymet purinnukleosidfosforylase (PNP), som også gir en bred defekt i både T-celler og B-celler (1). Noen virkelig effektiv behandling foreligger ikke ved denne formen for immunsviktsykdom, idet selv beinmargstransplantasjon med forlikelig giver oftest gir nokså dårlige resultater. Eksperimenter i cellekulturer har vist at overføring av genet til stamceller er mulig. En klinisk protokoll er utarbeidet og inklusjon av pasienter er startet, men ingen resultater foreligger.
Primær immunsvikt-sykdommer med defekt fagocyttfunksjon
Flere primær immunsvikt-sykdommer skyldes gendefekter som fører til nedsatt funksjon av fagocytterende celler, dvs. monocytter, makrofager og nøytrofile granulocytter. Disse tilstandene fører ofte til alvorlige infeksjonsproblemer.
Kronisk granulomatøs sykdom
Kronisk granulomatøs sykdom omfatter en gruppe sykdomstilstander karakterisert ved defekt fagocytt-funksjon som ved alle former affiserer ulike komponenter av nikotinamidadeninnukleotidfosfat (NADPH)-oksidasesystemet i fagocytterende celler. Fagocyttene med denne defekten fagocytterer mikroorganismer normalt, men er ikke i stand til å drepe de fagocytterte mikrobene. Dette fører til residiverende og kroniske infeksjoner, først og fremst med visse bakterier og sopparter. Tilstanden debuterer vanligvis tidlig i småbarnsalderen. I tillegg til permanent profylakse med trimetoprim-sulfametoxazol behandles disse pasientene i dag med γ -interferon (tre subkutane injeksjoner ukentlig), som har en viss klinisk effekt uten å være kurativt. Et lite antall pasienter er blitt helbredet med beinmargstransplantasjon.
I alt fire forskjellige gendefekter er beskrevet som årsak til kronisk granulomatøs sykdom. Tre forskjellige kliniske behandlingsstudier pågår i dag, som hver tar sikte på genterapeutisk korreksjon av en av de fire kjente gendefektene. Fra en av disse studiene foreligger det preliminære resultater, som viser at genterapien har ført til en viss, om enn meget begrenset, produksjon av fagocytter med normalt gen som persisterer flere måneder etter behandlingen (8).
Leukocyttadhesjonsdefekt
Ved denne sjeldne primære immunsviktsykdommen foreligger det en defekt i et gen for et viktig adhesjonsmolekyl (CD18), noe som gir svikt i granulocyttfunksjonen og alvorlige infeksjoner. Beinmargstransplantasjon kan helbrede sykdommen, men er ikke alltid mulig. Kliniske forsøk med genterapi er i gang (2).
Andre primær immunsvikt-sykdommer
For enkelte primær immunsvikt-sykdommer er genterapi fortsatt ikke mulig, til tross for at gendefekten er kjent, fordi genets funksjon ennå ikke er tilstrekkelig kartlagt. Dette gjelder blant annet kjønnsbundet, recessiv agammaglobulinemi (Bruton-type) og Wiskott-Aldrichs syndrom.
Konklusjon
Ved en rekke av disse til dels meget alvorlige primær immunsvikt-sykdommene vil genterapi sannsynligvis være den eneste mulige kurative behandling. Ved enkelte av sykdommene foreligger i dag alternative behandlingsformer, men bare i de tilfellene hvor beinmargstransplantasjon er mulig, dreier det seg om en kurativ effekt. Der slik behandling ikke er mulig eller ikke har hatt den ønskede effekt, vil genterapi kunne være et verdifullt alternativ. Ved en del primær immunsvikt-sykdommer kan pasienten overleve mer eller mindre sykdomsfri med andre behandlingsformer, for eksempel immunglobulinterapi ved former for hypogammaglobulinemi, γ -interferonbehandling ved kronisk granulomatøs sykdom, stadig antibiotikanvendelse etc. Også i disse tilfellene kan genterapi bli et attraktivt terapeutisk alternativ, dels pga. bedret livskvalitet for pasientene, dels ut fra helseøkonomiske vurderinger, siden mange av de eksisterende terapiformer er meget kostbare. Det er derfor grunn til å tro at genterapi vil finne sin plass i behandlingen av en rekke primær immunsvikt-sykdommer i årene fremover.
Jeg takker konsulent Ellen Finsberg for hjelp ved utarbeiding av figuren i artikkelen.
