Persisterende neonatal hyperinsulinemisk hypoglykemi er en sjelden tilstand, som i Vest-Europa og Nord-Amerika har en anslått hyppighet på 1 : 50 000 fødsler (1). I områder med mye inngifte, slik som i Saudi-Arabia, kan insidensen være opptil 1 : 2 500. Sykdommen er viktig som årsak til alvorlig neonatal hypoglykemi med fare for hjerneskade hos spedbarn. Tilstanden skyldes forsterket og uregulert insulinsekresjon fra β -cellene i pancreas, i de fleste tilfeller på grunn av defekte K + -kanaler (2). Økt insulinvirkning in utero gir økt fødselsvekt og et utseende som minner om nyfødte barn av diabetiske mødre. Patologisk-anatomisk kan man skille mellom to former med henholdsvis nodulær og diffus affeksjon (3). I et større materiale er nodulær form funnet hos 40 % (4). Tidligere brukte man betegnelsen nesidioblastose om den diffuse formen (5), fordi det ble observert grupper av β -celler uten den typiske organisering i langerhanske øyer. Denne betegnelsen bør utgå fordi det kan sees et liknende bilde hos friske nyfødte (3, 6).
Hyperinsulinisme kan behandles med diazoxid, som hemmer insulinsekresjonen ved en virkning på K + -kanalen (2), men denne behandlingen svikter ofte (4, 7). Den behandling som i dag foretrekkes er derfor kirurgisk. Men dette forutsetter en eksakt preoperativ diagnose, idet den nodulære form kan helbredes ved fjerning av et mindre område av kjertelen, mens den diffuse form krever subtotal (90 %) pankreatektomi (4). Den diagnostiske kartlegging har også viktige genetiske implikasjoner, idet den diffuse formen arves autosomalt recessivt, mens den nodulære form viser ikke-mendelsk arv (8).
En fullstendig diagnostisk utredning av persisterende neonatal hyperinsulinisme kan i dag ikke utføres i Norge. Vi omtaler i denne artikkelen tre norske barn som med suksess ble utredet og behandlet ved Höpital des Enfants Malades i Paris. Denne historien er et argument for at enkelte høyt spesialiserte medisinske tjenester for barn bør sentraliseres internasjonalt.
Pasienter
Pasient 1 . En gutt, andre barn av friske og ubeslektede foreldre, ble født til termin (40 uker) med vekt 4 340 g og lengde 53 cm. Pga. terapiresistent neonatal hypoglykemi ble han tre dager gammel overflyttet fra barneavdelingen, Fylkessjukehuset i Haugesund, til Barneklinikken i Bergen. Det ble påvist hyperinsulinisme med s-insulin 34,5 mU/l ved en samtidig blodglukose på 0,5 mmol/l. Stabile normalverdier for blodglukose lot seg ikke etablere med hydrokortison, glukagon eller diazoxid. To måneder gammel ble han overflyttet til Höpital des Enfants Malades i Paris. Insulinsekresjonsstudier (tab 1) viste et patologisk område i caput pancreatis. Han ble operert ti uker gammel med fjerning av størstedelen av pancreashodet og anastomose mellom pancreas og jejunum. Ductus choledochus måtte klippes og ble suturert. Postoperativt oppstod det kolangitt og sepsis. Senere har forløpet vært ukomplisert, bortsett fra en kortere periode med kolikkpregede magesmerter. Han har vist normal fysisk og mental utvikling, og med normal glukosebelastningskurve og normale verdier for s-insulin og C-peptid tre og et halvt år etter operasjonen regnes han som helbredet for sin hyperinsulinisme.
|
Tabell 1 Hovedtrekkene for diagnostikk og behandling av persisterende neonatal hyperinsulinemisk hypoglykemi ved Höpital des Enfants Malades i Paris
|
|
Prosedyre
|
Kommentar
|
|
Korrekt diagnose
|
Forsikre seg om at hypoglykemien er permanent og ikke transitorisk (en måned)
|
|
Teste følsomheten for diazoxid
|
Dosen som brukes er 15 mg/kg/dag Diazoxidrespons er sett i tre av 45 tilfeller
|
|
Selektiv kateterisering av vener fra pancreas med målinger av insulin og C-peptid
|
Mens det molare forhold mellom c-peptid og insulin er nær 10 i perifert blod, nærmer dette seg 1 når prøvene tas i pancreasparenkymet
|
|
Kirurgi med peroperativ histopatologisk undersøkelse
|
Omfanget av pancreasreseksjonen bestemmes av histologien sammen med resultatet av den preoperative kateterisering
|
Pasient 2. En jente, tredje barn av friske og ubeslektede foreldre, ble født en uke over beregnet termin med vekt 4 860 g og lengde 53 cm. En eldre bror med fødselsvekt 5 300 g hadde hatt forbigående neonatal hypoglykemi. Hun hadde terapiresistent hypoglykemi og ble to uker gammel overflyttet fra barneavdelingen, Sentralsjukehuset i Møre og Romsdal, til Barneklinikken i Bergen. Det ble funnet forhøyet s-insulin, med 23,2 mU/l ved en samtidig blodglukose på 2,7 mmol/l. Behandlingsforsøk med hydrokortison, glukagon, diazoxid og nifedipin førte ikke frem. Seks uker gammel ble hun overflyttet til Höpital des Enfants Malades i Paris, hvor hun ble undersøkt og behandlet etter standard protokoll (tab 1). Det ble påvist diffuse forandringer i den endokrine pancreas, og hun ble operert med subtotal (90 %) pankreatektomi. Ved kontroll to og et halvt år etter operasjonen hadde hun en diabetiskpreget glukosebelastningskurve uten tegn til klinisk diabetes. Hun har samtidig tendens til fastende hypoglykemi og får derfor hyppige måltider, men ingen spesiell behandling. Psykomotorisk har hennes utvikling vært normal.
Pasient 3. En jente, første barn av friske og ubeslektede foreldre, ble forløst med keisersnitt på grunn av mekaniske misforhold. Vekt og lengde var henholdsvis 4 560 g og 52 cm. Ved Rikshospitalet ble det de første levedager registrert hypoglykemi (1,5 – 2 mmol/l). Ni dager gammel ble hun overflyttet til Barnesenteret, Ullevål sykehus. Her stabiliserte blodglukosen seg (2,5 – 3,5 mmol/l), og hun ble utskrevet etter 11 dager. Hjemme ble hun fullammet med hyppige måltider, men hadde et kortvarig krampeanfall. Fem og en halv måned gammel ble hun syk med feber, uro og senere kramper. Ved ny innleggelse var hun klinisk upåfallende, men hadde blodglukose på 1,4 mmol/l. S-insulin og C-peptid var innenfor referansegrensene, men ratio insulin/glukose var patologisk (84/1,7). Barnet hadde vedvarende behov for glukose intravenøst i tillegg til peroral ernæring, for å holde blodglukose over 3,0 mmol/l. I en periode trengte hun glukose 200 mg/ml intravenøst. Barnet utviklet spisevegring og måtte etter hvert sondeernæres i tillegg til intravenøs glukose. Langsomt absorberbare karbohydrater peroralt hjalp noe. Behandling med diazoxid førte ikke frem. Sju måneder gammel ble hun overflyttet til Höpital des Enfants Malades i Paris, hvor hun ble undersøkt etter standard protokoll (tab 1). Det ble funnet en fokal hyperinsulinisme og det ble utført partiell reseksjon av pancreas. Det postoperative forløp var ukomplisert. Mer enn to år senere viser hun normal utvikling og har ingen tegn til hypoglykemi.
Diskusjon
Undersøkelse og behandling av barn med persisterende neonatal hyperinsulinemisk hypoglykemi krever medvirkning av en rekke fagpersoner med særlig kompetanse og interesse (obstetriker, pediater, røntgenolog, genetiker, endokrinolog, kirurg og patolog). Det er en sjelden og ressurskrevende tilstand som reiser spørsmål om sentralisert service.
De tre omtalte spedbarna hadde en alvorlig grad av hyperinsulinemisk hypoglykemi som ikke lot seg kontrollere ernæringsmessig eller medikamentelt. For å sikre et optimalt tilbud av videre utredning og kirurgisk intervensjon ble barna henvist til Höpital des Enfants Malades i Paris etter at folketrygden hadde gitt de nødvendige garantier. Ved dette sykehuset er det utviklet en undersøkelsesteknikk som er unik, ved å kunne skille mellom sykdommens fokale og diffuse former med stor treffsikkerhet (4). Dette er meget viktig fordi partiell reseksjon ved den fokale form kan være helbredende (4), mens prognosen ved subtotal pankreatektomi er usikker, med komplikasjoner som veksthemning, residiv av hypoglykemi eller utvikling av diabetes (7, 9, 10).
Skillet mellom diffus og fokal form har også genetiske implikasjoner, siden den diffuse form arves autosomalt recessivt eller autosomalt dominant (11, 12), mens den fokale form viser ikke-mendelsk arv (8, 13) (tab 2). Denne siste subtypen viser et interessant arvemønster som krever to genetiske hendelser. Første hendelse er når barnet arver en mutasjon som er oppstått i et paternelt SUR1- eller KIR6.2-allel. Denne mutasjonen finnes i arvestoffet i alle cellene hos barnet. Den neste hendelse er et spesifikt, somatisk tap av maternelle alleler i form av delesjon på kromosom 11p15 bare i den fokale lesjonen i pancreas. Herved demaskeres den paternelle heterozygositet.
|
Tabell 2 Ulike former for kongenitt hyperinsulinisme 1
|
|
Arvegang
|
Kromosom
|
Gener
|
Funksjonell defekt
|
Morfologi
|
Klinikk
|
|
Autosomal recessiv
|
11p15.1
|
SUR1 eller KIR6.2
|
K * -kanal Tap av funksjon Blokkering av K + -transport ut av cellen medfører økt kalsiuminfluks og dermed økt insulinsekresjon
|
Diffus
|
Ca. 50 rapporterte tilfeller Høy fødselsvekt Alvorlig Ufølsom for diazoxid
|
|
Ikke-mendelsk
|
11p15
|
SUR1, KIR6.2 og andre gener i denne regionen
|
K * -kanal, se over Somatisk delesjon av maternelle gener i regionen 11p15 i selve tumor samtidig med kimcellemutasjon av paternelt SUR1 eller KIR6.2
|
Fokal
|
Hyppig (30 – 50 %) Mild Følsom for diazoxid Spontan remisjon?
|
|
Autosomal dominant
|
10q23.3 eller 7p15-p13
|
GLUD-1 eller GCK
|
Glutamatdehydrogenase eller Glukokinase Økt funksjon som gir økt insulinsekresjon
|
Diffus
|
Ca. 40 kjente tilfeller Mild Følsom for diazoxid
|
|
|
Patofysiologien ved persisterende neonatal hyperinsulinisme er i dag langt på vei klarlagt som en K + -kanaldefekt (2). Betacellen i pancreas har en ATP-sensitiv K + -kanal som er nær knyttet til en sulfonylureareseptor i en funksjonell enhet (fig 1) (14). Mutasjoner i to gener (SUR1 og KIR6.2) som koder for denne enheten, leder til blokk av K + -kanalen (15, 16). Dette medfører depolarisering av cellemembranen, influks av Ca ++ og økt frisetting av insulin. Diazoxid hemmer insulinfrisettingen ved å åpne K + -kanalen.
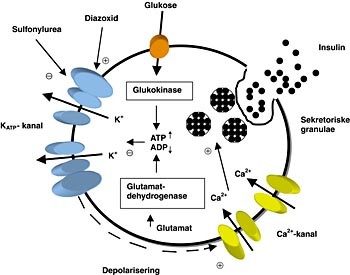
Figur 1 Reguleringen av insulinsekresjonen i β -cellen i pancreas. Defekter i glukokinase, glutamatdehydrogenase eller en av de to subenhetene i K + -kanalen leder alle til redusert transport av K + ut av β -cellen. Dette gir depolarisering av cellemembranen, influks av Ca 2+ og økt insulinsekresjon. Figuren er modifisert etter Ashcroft (14)
Höpital des Enfants Malades mottar pasienter fra store deler av Sør- og Mellom-Europa, og har med hell behandlet tre norske barn med hyperinsulinisme, som her omtalt. To av barna ansees som helbredet, mens det hos den tredje på lengre sikt er en risiko for utvikling av diabetes etter subtotal pankreatektomi.
Det er tvilsomt om man for en tilstand som dette bør bygge opp en komplett nasjonal ekspertise. I Paris kan man etter få år gjennomgå og etterundersøke behandlingsserier med 25 – 30 pasienter av den aktuelle type. I Norge vil man i gjennomsnitt se en pasient i året.
Når skal barn sendes til behandling i utlandet?
Dette er et spørsmål av generell karakter som belyses av de presenterte kasuistikker. Spørsmålet, som må vurderes fortløpende ut fra aktuelle undersøkelses- og behandlingstilbud hjemme og ute, har også prinsipielle sider. Bruk av internasjonal ekspertise i undersøkelsen av sjeldne pediatriske sykdomstilstander er en veletablert praksis i vårt land, og er ofte basert på personlige faglige kontakter. Vanligvis sendes biologisk materiale i form av blod, serum, vevsprøver eller dyrkede celler. Nødvendigheten av å sende spesielle prøver er uomtvistelig. Mer diskutabelt er det om og når pasienten skal sendes. Motargumenter vil her være de språklige og kulturelle barrierer som familiene møter, og nærheten til hjemmemiljøet som går tapt. Medisinsk kan det også bety et tap, idet faglige utfordringer er stimulerende, og utredning av sjeldne tilstander kan ha positive ringvirkninger i en videre sammenheng. Men det avgjørende argument for i spesielle tilfeller å sende barn til behandling i utlandet må selvsagt være medisinsk, ved at barn med spesielle lidelser kan få optimal behandling ved høyt spesialiserte sentre der man har stort erfaringsgrunnlag.
