Den sentrale godkjenningsprosedyre er obligatorisk for legemidler fremstilt ved en av tre nærmere spesifiserte bioteknologiske metoder. I tillegg har man åpnet for at prosedyren kan benyttes på frivillig basis for legemidler som ansees å ha en vesentlig innovativ verdi, terapeutisk interesse, for legemidler fremstilt av blod eller plasma og for legemidler som inneholder et nytt aktivt virkestoff.
Ved sentral prosedyre søker et legemiddelfirma The European Agency for the Evaluation of Medicinal Products (EMEA) om markedsføringstillatelse i samsvar med de gjeldende krav til dokumentasjon, og får ved positiv vurdering markedsføringstillatelse som gir tilgang til alle EU-land samt Norge og Island via EØS-avtalen. Innen 30 dager etter endelig vedtak i EU-kommisjonen må Statens legemiddelverk fatte sammenfallende vedtak. Deretter må pris fastsettes, og legemidlet kan dermed markedsføres på det norske markedet.
Arbeidet i komiteen for legemidler til mennesker
Sentralt i utredningen av nye legemidler i sentral prosedyre står en vitenskapelig legemiddelkomité, komiteen for legemidler til mennesker (Committee for Proprietary Medicinal Products, CPMP). Komiteen er knyttet til EMEA, som koordinerer komitéarbeidet og tilrettelegger bruk av vitenskapelig ekspertise for CPMP (fig 1). Komiteen består av to medlemmer fra hvert medlemsland, inkludert Norge og Island. Fra Norge har det vært ansatte ved Legemiddelverket som har vært nominert som CPMP-medlemmer. Komiteen møtes 3 – 4 dager en gang hver måned på kontoret for legemiddelvurdering (EMEA), som er lokalisert i London.
Ved utredningen av den enkelte søknad for et nytt legemiddel utnevnes en rapportør og en medrapportør blant medlemmene i komiteen for legemidler. Legemiddelmyndighetene i rapportørs og medrapportørs hjemland utfører sin uavhengige vurdering av alle delene av søknaden, inkludert farmasøytisk-kjemisk, preklinisk og klinisk dokumentasjon, og utarbeider forslag til beslutning. Utredningen skal være ferdig innen 210 kalenderdager. Under behandlingsprosessen vil legemiddelfirmaet skriftlig besvare spørsmål og komplettere eventuelle mangler i sin søknad, men den tiden firmaet bruker på dette, inngår ikke i tidsrammen for behandlingstid (klokken stopper).
Forslaget til beslutning diskuteres i flere omganger i CPMP før endelige råd foreligger ved dag 210. Det er ofte nødvendig med assistanse fra ledende spesialister til å vurdere den kliniske effekt og klinisk relevans i relasjon til sikkerhet av nye legemidler. Det er også ofte nødvendig med bistand fra eksperter ved indikasjonsutvidelser og sikkerhetsvurderinger av allerede godkjente legemidler.
I forbindelse med de faglige diskusjonene i komiteen, som i hovedsak er relatert til kliniske problemstillinger, kan medlemmene bringe med seg eksperter fra sine hjemland for å bistå i faglige diskusjoner. På denne måten kan de kliniske miljøene i medlemslandene være med på å påvirke prosessene. Komiteens beslutning er egentlig et råd til EU-kommisjonen, som er den endelige beslutningsinstans. EU-kommisjonen har til nå fulgt komiteens råd i slike saker.
Komiteen har en rekke faste arbeidsgrupper samt ad hoc-arbeidsgrupper som arbeider med overordnede retningslinjer i forhold til dokumentasjon av nye legemidler og andre faglige problemstillinger (fig 1). Dette arbeidet danner en vesentlig basis for den kliniske utvikling av nye legemidler. På EMEAs nettside (1) finnes det oppdaterte preparatomtaler for samtlige godkjente produkter i sentral prosedyre. Mer utførlige redegjørelser for beslutningsgrunnlaget og beslutningsprosessen for disse produkter finnes i form av såkalte European Public Assessment Reports (EPAR) (1).
Gjensidig anerkjennelsesprosedyre
I gjensidig anerkjennelsesprosedyre (mutual recognition procedure) kan et legemiddelfirma selv velge hvilke land det ønsker markedsføringstillatelse i. Firmaet søker først i ett land. Hvis legemidlet godkjennes der, ber firmaet om at denne første utredningen og markedsføringstillatelsen anerkjennes i ett eller flere andre EØS-land der firmaet ønsker å markedsføre sitt preparat. Dersom et av disse andre landene har alvorlige innvendinger, kan det resultere i at den første markedsføringstillatelsen må trekkes tilbake etter en voldgiftsprosedyre i CPMP. En annen mulighet er at søker trekker søknaden i det landet som har innvendinger.
En oversikt over alle legemidler som er godkjent i denne prosedyren, finnes på egen Internett-adresse (2). I forbindelse med revisjonen av EUs prosedyrer foreligger det et forslag om å begrense gjensidig anerkjennelsesprosedyre til generika, men det er ikke enighet i medlemslandene om dette.
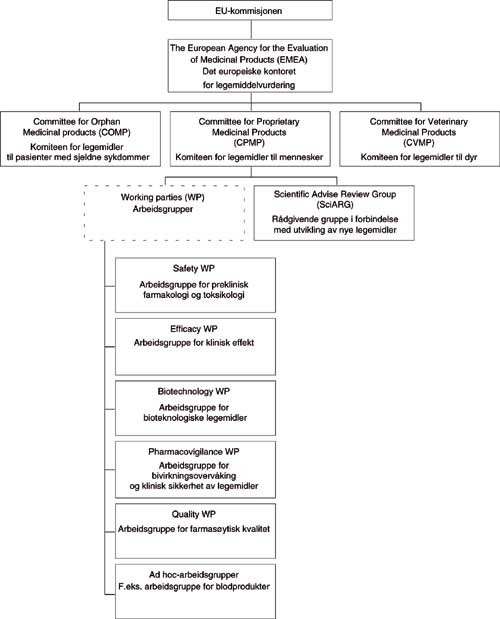
Figur 1 Oversikt over sentral legemiddelforvaltning inkludert komiteer og arbeidsgrupper i EU/EØS
Nasjonale søknader
En ren nasjonal godkjennelse er som hovedregel kun mulig for nye administrasjonsformer eller styrker av allerede markedsførte legemidler i Norge, eller dersom legemidlet kun skal markedsføres i Norge. Naturlegemidler godkjennes i denne prosedyren.
Statens legemiddelverk
Grunntanken bak den sentrale godkjenningsprosedyre har vært at sikre og effektive, innovative legemidler raskt skal bli tilgjengelige i samtlige EU/EØS-land. En annen viktig målsetting har vært å skape identisk preparatomtale og pakningsvedlegg i EU/EØS-området. Med hensyn på ulikhetene i Europa når det gjelder sykdomspanorama, terapitradisjoner, informasjon til forskrivere og pasienter etc., har det vært overraskende stor andel av avgjørelsene som fattes i konsensus eller nesten konsensus.
Statens legemiddelverk har fra 1.1. 2000 vært representert med to medlemmer i komiteen for legemidler til mennesker (CPMP). Dessuten har man representanter i komiteens arbeidsgrupper. Det har tatt tid å bli kjent med et stort, nytt system, men erfaringen så langt er god. Selv om våre stemmer som EØS-medlemmer ikke teller med ved formelle avgjørelser, kan vi påvirke avgjørelsene. Dette skyldes at faglige innspill under saksgangen og diskusjonen i komiteen er styrende for de vedtak som fattes. I særlig grad påvirker man beslutningsprosessene når man påtar seg utredningsoppgaver, for eksempel som rapportør for nye legemiddelsøknader. Dette krever faglig ekspertise på høyt nivå, og det er ønskelig med et sterkere samarbeid mellom de kliniske miljøene og Statens legemiddelverk for å oppnå dette.
Norge har deltatt aktivt i flere arbeidsgrupper, og har vært spesielt aktive på vaksineområdet. Vi har også vært involvert i arbeidet som endte opp med en standpunktserklæring vedrørende forskjell på annen og tredje generasjons p-piller. Det er gode muligheter til å påvirke prosesser på mange fagområder, men dette krever engasjement og faglig tyngde.
Norge har så langt vært oppnevnt som medrapportør ved tre utredninger av legemidler til mennesker. Erfaringene med dette arbeidet er gode, og våre utredninger har vært fullt på høyde med andre lands. Statens legemiddelverk ønsker flere slike oppdrag, da dette også vil gi bedre kompetanse og være en viktig inntektskilde.
Legemiddelverkets rolle i forhold til EU-avgjørelsene blir også å informere norske leger om nye legemidler og legemidlenes plassering i det terapeutiske bilde. Det er ønskelig at de kliniske miljøene i Norge har forståelse for muligheten til å påvirke prosesser som til slutt kan være viktige for folkehelsen. Rekruttering av leger til dette spennende og utfordrende arbeidet ved Legemiddelverket har vært vanskelig.
Spalten er redigert av Olav Spigset i samarbeid med Avdeling for legemidler ved St. Olavs Hospital og de øvrige klinisk farmakologiske miljøene i Norge.
Se også kunnskapsprøve på www.tidsskriftet.no/quiz
