Det er gått over 100 år siden det første kontrollerte kliniske forsøket. I 1898 undersøkte dansken J. Fibiger effekt av serumbehandling ved difteri. Pasienter som ble innlagt den ene dagen, fikk serumbehandling, mens de som ble innlagt neste dag, ikke fikk behandling. Forsøket varte i ett år og omfattet 484 pasienter. Åtte pasienter i behandlingsgruppen døde, mot 30 i den like store kontrollgruppen. Interessen for disse forsøkene skjøt først fart etter den annen verdenskrig, og i 1948 gjennomførte UK Medical Research Council en studie av streptomycinbehandling ved tuberkulose med et randomisert forsøk som er blitt en milepæl i medisinsk forskning (1).
De fleste kjenner begrepene «randomisert» og «dobbeltblind» og vet at disse er essensielle elementer i en god klinisk utprøvning. Vi vil i denne artikkelen belyse en del av hovedprinsippene for randomiserte kliniske forsøk og begrunne hvorfor de forskjellige prosedyrene er viktige.
Erfaringer fra en studie som en av oss (ML) deltok i, vil bli benyttet for å illustrere hvilke problemer som kan oppstå i praksis og hvordan man kan møte dem (2). Dette var en sammenliknende studie av effekten av penicillin V, amoksicillin og placebo ved akutt sinusitt hos pasienter i allmennpraksis hvor diagnosen ble bekreftet med CT-undersøkelse. Studien var randomisert og dobbeltblind.
Hvorfor randomisere?
Hensikten med å randomisere pasienter til forskjellig type behandling er å sikre en rettferdig sammenlikning av behandlingene. Metoden brukes for å oppnå tilfeldig fordeling av pasienter til behandlingsgruppene slik at ukjente faktorer som kan tenkes å påvirke forløpet av sykdommen, blir tilfeldig fordelt i gruppene.
Hvis man studerer effekten av et medikament, for eksempel et smertestillende middel, er det svært uheldig hvis alle med sterke smerter kommer i den ene gruppen og de med moderate i den andre. Vi vil ønske en lik fordeling av grad av smerte i de to gruppene og gjerne også omtrent lik fordeling av kjønn, alder og grunndiagnose. Randomisering sørger for at alle faktorer som kan påvirke sykdomsforløpet, både kjente og ukjente, fordeles tilfeldig i de to gruppene. Randomisering er ikke nødvendigvis noen garanti for at fordelingen av prognostiske faktorer i gruppene blir lik, men i store forsøk vil en tilfeldig fordeling vanligvis ikke medføre skjevheter av betydning. I mindre studier kan en lik fordeling sikres for eksempel med stratifisering og blokkrandomisering.
Pasientene deles da inn i undergrupper (strata), og det lages egne randomiseringslister for hvert stratum. På denne måten sørger man for balanse slik at like mange pasienter i hver undergruppe får hver av behandlingene. Det er fornuftig å begrense antall stratifiseringsvariabler til pasientkarakteristika man vet har betydning for utfallet. Aktuelle faktorer kan være alder, kjønn eller sykdommens alvorlighetsgrad. Det er verdt å merke seg at alle stratifiseringsvariabler som er benyttet i randomiseringsprosessen, også skal være med i den statistiske analysen.
Tabell 1 viser et eksempel på randomisering av pasienter i to strata. Med en blokkstørrelse på tre vil det for hver tredje pasient som inkluderes i et stratum være like mange som har fått hver av de tre aktuelle behandlingene. For å redusere risikoen for at behandlende lege skal kunne gjette hvilken behandling neste inkluderte pasient vil få, vil man ofte benytte en blokkstørrelse som er 2 – 4 ganger så stor som antall behandlingsgrupper eller som varierer fra blokk til blokk. I sinusittstudien ble det i tillegg stratifisert etter resultat av CT-funn inndelt i tre grupper (ensidig eller tosidig maxillarissinusitt eller sinusitt i et av de andre bihuleområdene). I kombinasjon med skåre var det altså i praksis seks forskjellige strata.
Hvorfor blinde?
Blinding er nødvendig for å sikre at registrering og fortolkning ikke påvirkes av subjektive antakelser om effekt av behandling. Vanligvis pakkes medikamenter ferdig etter en computergenerert randomiseringsliste. Pakninger og innhold ser identiske ut, og det eneste som skiller dem fra hverandre, er et pasientnummer. En slik studie er dobbeltblind fordi verken lege eller pasient vet hvilken behandling pasienten får. På den måten unngår man at pasientens eller behandlende leges subjektive oppfatninger om behandlingseffekt påvirker pasientens respons på legemidlet.
Det er dokumentert at dersom pasient eller lege vet hvilket medikament pasienten har fått, vil dette påvirke pasientens angivelse av symptomer og legens tolking av resultatene (3). Det er også vist at det å ikke skjule prosedyren for fordeling til behandling eller kontroll ved rekruttering til randomiserte forsøk, ofte resulterer i en skjev fordeling av prognostiske faktorer (seleksjon) (4). Dermed kan man få estimater for effektstørrelse som er større og oftere statistisk signifikante enn de estimatene man beregner når fordelingsprosedyren er skjult. I studier hvor intervensjonen ikke er medikamentell, vil dobbeltblinding som regel være umulig. Et eksempel fra Norge kan være bruk av skjedeinnlegg for å styrke bekkenmuskulaturen ved inkontinens (5). I slike tilfeller er det en fordel om randomiseringskoden ikke brytes før etter at statistiske analyser er utført.
Signifikanstest
For å sammenlikne effekten av forskjellige behandlinger benytter man ofte en signifikanstest. Hvis den såkalte p-verdien blir lav, er det uttrykk for at det er lite sannsynlig at den observerte forskjellen i effekt av behandlinger skyldes tilfeldighet. Tradisjonelt regnes en forskjell mellom to behandlinger som statistisk signifikant når p < 0,05, dvs. når det er mindre enn 5 % sannsynlighet for at den forskjellen som er observert (eller en enda større forskjell), skyldes tilfeldighet og ikke er uttrykk for en reell forskjell. Når man finner en statistisk signifikant effekt i et forsøk, betyr det altså at det er sannsynliggjort at det er en reell effekt. Men samtidig er en høy p-verdi ikke dokumentasjon av at to behandlinger har lik effekt. Det er fornuftig ikke å se seg blind på den «magiske» 5 %-grensen for statistisk signifikans, men rapportere beregnede p-verdier og sette dem i en større sammenheng for å vurdere effekten av et nytt behandlingsregime.
|
Tabell 1 Eksempel på stratifisering og blokkrandomisering for en sinusittstudie med to strata og en blokkstørrelse på 3. A: amoksicillin, B: penicillin V, C: placebo
|
|
Klinisk skåre < 9
|
Klinisk skåre ≥ 9
|
|
A
|
B
|
|
C
|
C
|
|
B
|
A
|
|
|
|
C
|
B
|
|
B
|
A
|
|
A
|
C
|
|
|
|
C
|
A
|
|
A
|
B
|
|
B
|
C
|
|
·
|
·
|
|
·
|
·
|
|
·
|
·
|
Effektestimater
For å bedømme effekt av en behandling er det ikke nok bare å angi en p-verdi. Behandlingseffektens størrelse må også estimeres. I tillegg angis et 95 % konfidensintervall som er et uttrykk for usikkerheten i estimatet av behandlingseffekt. Jo smalere konfidensintervallet er, desto mer presist er effekten estimert. Store forsøk vil gi presise estimater, mens små forsøk resulterer i vide konfidensintervaller og stor usikkerhet med hensyn til sann effekt av behandling.
Ekvivalens
For dokumentasjon av at to behandlinger har like god effekt (ekvivalens), er det ikke tilstrekkelig at man ikke finner statistisk signifikant forskjell mellom dem. Antall pasienter som er inkludert i en studie, vil påvirke p-verdien. Med få pasienter vil det være nesten umulig å oppdage selv store forskjeller mellom behandlinger. Studier der man ikke finner statistisk signifikant forskjell mellom behandlinger, refereres noen ganger til som «negative studier». Dette uttrykket er uheldig fordi det synes å indikere at studien har vist at det ikke er forskjell mellom to behandlinger, mens det som vanligvis er tilfellet, er at det ikke er vist forskjell. De to utsagnene er ikke like. Manglende dokumentasjon av effekt er ikke det samme som dokumentasjon av manglende effekt (6)!
I praksis vil man vurdere hvorvidt to behandlinger er like gode ved hjelp av 95 % konfidensintervaller. Man bør på forhånd definere den største forskjellen man vil akseptere for likevel å kunne betrakte de to behandlingene som ekvivalente. Dersom et 95 % konfidensintervall for forskjell har en øvre grense som er mindre enn den forhåndsdefinerte forskjellen, kan dette betraktes som at man har vist at den nye behandlingen ikke er dårligere (non-inferiority).
I sinusittstudien var andelen pasienter som ble friske eller mye bedre etter ti dagers behandling 32/39 (82 %) i penicillin V-gruppen og 39/44 (89 %) i amoksicillingruppen; 95 % konfidensintervall for forskjell (–8 %, 22 %). En mulig 22 % forskjell kan neppe ansees som dokumentasjon av at de to antibiotikaregimene er like gode. Med et større antall pasienter inkludert ville konfidensintervallet blitt smalere, og en klar konklusjon kunne lettere vært trukket.
Undersøkelsens styrke
En viktig del av planleggingen av en legemiddelutprøvning består i å anslå hvor mange pasienter som skal inkluderes. Dette gjøres vanligvis ved at man definerer den minste forskjellen mellom to behandlinger som det vil være betydningsfullt å oppdage (klinisk relevant forskjell), bestemmer ønsket sannsynlighet for å oppdage denne (teststyrke eller «power», ofte satt til 80 % eller 90 %) og ut fra disse valgene beregner hvor mange som bør inkluderes.
I sinusittstudien ble styrkeberegningen basert på tidligere kjente data om varighet av en sinusittepisode, i gjennomsnitt ti dager. Vi kjente variasjonen innenfor gruppen (SD = 3,4), og ønsket var å kunne oppdage forskjell mellom behandlingsgruppene dersom den reelle forskjellen i varighet var minst 20 %, dvs. to dager. Dette medførte at vi skulle ha tre grupper på 60 pasienter for å oppnå 90 % sannsynlighet for å finne en slik forskjell. Da vi hadde rekruttert pasienter i to vintre, hadde vi 45 pasienter i hver gruppe og besluttet å avslutte studien, selv om vi ikke hadde oppnådd det pasientantall som på forhånd var planlagt. Det ble vist signifikant forskjell mellom placebo og hver av de to antibiotikaene. Det ble derimot ikke vist signifikant forskjell mellom penicillin V og amoksicillin, men som nevnt tidligere er likhet ikke dermed dokumentert.
Beregning av antall pasienter (ramme 1)
En rekke programpakker til beregning av utvalgsstørrelse er tilgjengelig, men for sammenlikning av to (eller flere) uavhengige utvalg kommer man langt ved hjelp av to enkle formler. Formlene som gis nedenfor passer ikke når man har flere observasjoner fra samme pasient, for eksempel i overkrysningsstudier.
For målevariabler kan man beregne utvalgsstørrelse ved hjelp av følgende formel:
der n står for antall pasienter i hver gruppe, SD er standardavviket til observasjonene, D den forskjellen man ønsker å avdekke dersom den finnes (klinisk relevant forskjell) og k en konstant som avhenger av valgt signifikansnivå og teststyrke. De mest alminnelige valg er en tosidig test på 5 %-nivå og teststyrke 80 % eller 90 %. De to kombinasjonene gir henholdsvis k = 7,9 og k = 10,5. Jo høyere teststyrke (og jo lavere signifikansnivå) man ønsker, desto større blir k og dermed antall pasienter.
I sinusittstudien ble D satt til to dager, SD var basert på tidligere studier anslått til 3,4, og med valgt signifikansnivå 5 % og teststyrke 90 % fikk man k = 10,5 . Setter man disse tallene inn i formelen, finner man n = 60 pasienter i hver gruppe.
Et problem man straks støter på når man skal estimere det antall pasienter det er nødvendig å inkludere i en studie, er å finne et godt anslag for standardavviket. Det sanne standardavviket er ukjent, og man blir nødt til å basere seg på tidligere studier eller utføre en pilotstudie med for eksempel ti pasienter for å sikre seg at man benytter et rimelig anslag. Er det virkelige standardavviket større enn man på forhånd antok, vil man inkludere for få pasienter og dermed kunne gå glipp av en forskjell som kanskje kunne ha stor klinisk interesse.
Når man vil sammenlikne to grupper der effektvariabelen er binomisk (ja/nei), er følgende formel nyttig:
hvor n igjen er antall pasienter i hver gruppe, p ₁ er andel «ja» i gruppe 1, p ₂ er andel «ja» i gruppe 2, og k avhenger av valgt signifikansnivå og teststyrke, som i formelen for målevariabler. Dersom andelen pasienter som har respons på behandling i kontrollgruppen på forhånd antas å være p ₂= 0,6 og man ønsker 90 % teststyrke for å oppdage en absolutt økning i respons på 20 % (svarende til p ₁= 0,8 og k = 10,5 med tosidig signifikansnivå 5 %), må n = 105 pasienter inkluderes i hver gruppe.
Selv om teststyrken blir høyest dersom det totale pasientantallet fordeles på grupper med omtrent like mange pasienter i hver, trenger behandlingsgruppene ikke nødvendigvis å være like store. I praksis taper man lite med hensyn til teststyrke selv om fordelingen på to grupper er såpass skjev som 2 : 1.
Tips til utdypende lesning om kontrollerte kliniske studier er gitt i ramme 2.
Intern validitet
Alle de forholdene som er knyttet til gjennomføringen av en klinisk behandlingsstudie, kan oppsummeres som intern validitet. De viktigste typer skjevhet (bias) er vist i figur 1 (7). Randomisering og blinding benyttes for å unngå disse problemene og dermed sikre at en klinisk studie har høy intern validitet.
Ramme 1
Formler for beregning av utvalgsstørrelse, uavhengige utvalg
Symbolene er forklart i teksten.
Formlene passer ikke når man har par av observasjoner på samme pasient.
Ramme 2
Anbefalt utdypende litteratur om kontrollerte kliniske forsøk
-
Pocock SJ. Clinical trials. A practical approach. New York: Wiley, 1983.
-
Sackett DL, Haynes RB, Guyatt GH, Tugweel P. Clinical epidemiology – a basic science for clinical medicine. Boston: Little, Brown, 1991.
-
www.emea.eu.int/pdfs/human/ich/036396en pdf (1.10.2002)
Ekstern validitet
I en klinisk utprøvning inkluderer man et utvalg av pasienter, og på grunnlag av dette forsøker man å trekke slutninger om en populasjon. For at disse slutningene skal være gyldige, må utvalget av pasienter være representativt for den populasjonen som i fremtiden vil få den aktuelle behandlingen.
Dersom man vil sikre at pasientgruppen har en bestemt sykdom, for eksempel akutt bakteriell sinusitt, og bruker en referansestandard som røntgen eller CT av bihuler for å sikre dette, slik det ble gjort i vår studie, blir resultatet ikke nødvendigvis generaliserbart. Man har oppnådd en «ren gruppe» hvor man kan utprøve effekten av antibiotika mot placebo. I sinusittstudien ble i alt 244 pasienter med mistenkt sinusitt i utgangspunktet henvist fra allmennpraktiker. Kun 130 av disse fikk påvist sinusitt ved CT og ble dermed inkludert i studien. Resultatene fra studien kan bare sies å være relevante for 55 % av utgangspopulasjonen. I klinisk allmennpraksis kan vi ikke bruke slike diagnostiske metoder hver gang vi har en pasient med mistenkt klinisk sinusitt, vi vil basere oss på kliniske symptomer, tegn og blodprøver som har en relativt stor usikkerhet. Dermed vil populasjonen vanligvis være en annen enn den som ble undersøkt i forsøket, og erfaringene fra det kontrollerte kliniske forsøket kan ikke nødvendigvis overføres direkte.
Et annet vesentlig spørsmål for om de rekrutterte pasientene er representative, er hvor stor andel av mulige kandidater som blir rekruttert til studien. Hvis de rekrutterende legene klarer å rekruttere 70 % av mulige kandidater til studien, kan vi være langt sikrere på at pasientene speiler den kliniske virkeligheten enn hvis de bare klarer å rekruttere 10 %.
Et siste mål for kvalitet på en studie er at frafallet bør være lavt, ellers kan det bli vanskelig å tolke resultatene av studien. For at studien skal være valid, må den ikke være beheftet med eksklusjonsskjevhet. Pasienter som trekker seg underveis i en studie og ikke fullfører, bør ideelt være likt fordelt på de to behandlingsgruppene. I en placebokontrollert studie av antibiotika ved luftveisinfeksjoner kan det være en fare for at pasienter som får placebo, vil få en høyere frekvens av frafall og at flere med alvorligere sykdom vil falle fra enn i behandlingsgruppen. Dette kan gjøre resultatene vanskelige å tolke.
Også i den statistiske analysen må man tenke gjennom hvilken populasjon av pasienter som skal inngå. Vanligvis baserer man analysen på det såkalte behandlingsintensjonsprinsippet som innebærer at alle randomiserte pasienter skal inkluderes i den statistiske analysen, enten de ble behandlet slik de ble randomisert eller ikke. På denne måten unngår man systematiske skjevheter og overestimering av forskjell mellom behandlinger (8).
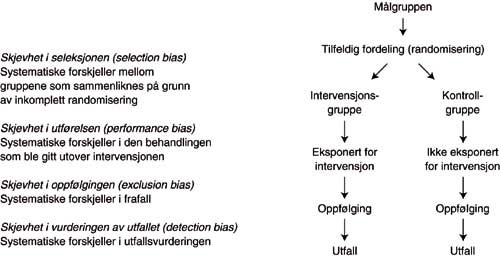
Figur 1 Kilder til skjevhet i et randomisert forsøk (seleksjon, utførelse, eksklusjon og vurdering). Omarbeidet etter (7)
|
Tabell 2 Presentasjon av effektforskjell i to ulike studier
|
|
|
Resultat (%)
|
|
|
|
|
Problemstilling
|
Effektmål
|
Behandling
|
Kontroll
|
Absolutt forskjell (%)¹
|
Relativ forskjell (%)¹
|
NNT
|
|
Amoksicillin versus placebo ved sinusitt (2)
|
Friske/mye bedre dag 10
|
89
|
56
|
33 (89 – 56)
|
59 (33/56)
|
3 (100/33)
|
|
Warfarin versus placebo ved atrieflimmer (9)
|
Slag
|
1,4
|
4,5
|
3,1 (4,5 – 1,4)
|
69 (3,1/4,5)
|
32 (100/31)
|
|
Tabell 3 Sjekkliste for rapportering av randomiserte kliniske forsøk (CONSORT-statement) (13)
|
|
Artikkeldel og tema
|
Punkt
|
Beskrivelse
|
|
Tittel og sammendrag
|
1
|
Hvordan deltakere ble fordelt til intervensjoner («randomisert»)
|
|
Introduksjon/bakgrunn
|
2
|
Vitenskapelig bakgrunn og forklaring av begrunnelse for studien
|
|
Metoder
|
|
|
|
Deltakere
|
3
|
Hvilke pasienter som kunne rekrutteres, og hvor og hvordan data ble innsamlet
|
|
Intervensjoner
|
4
|
Presis beskrivelse av intervensjonen som skulle gis til hver gruppe og hvordan den faktisk ble administrert
|
|
Målsetting
|
5
|
Spesifikke målsettinger og hypoteser for studien
|
|
Effektmål
|
6
|
Klart definerte primære og sekundære effektmål, og, hvis aktuelt, metoder brukt for å sikre datakvalitet (trening av observatører etc.)
|
|
Studiens størrelse
|
7
|
Hvordan studiens størrelse ble bestemt med styrkeberegning, og, hvis aktuelt, beskrivelse av interimanalyser og regler for å stoppe studien
|
|
Randomisering
|
|
|
|
Sekvensbestemmelse
|
8
|
Metode brukt for å generere randomiseringssekvenser, inkludert detaljer om begrensninger (blokkrandomisering, stratifisering)
|
|
Forseglet allokering
|
9
|
Metode brukt ved praktisk utføring av randomisering (f.eks. nummererte medisinbokser eller sentral telefon) og om sekvensen var forseglet inntil pasienten ble inkludert
|
|
Implementering
|
10
|
Hvem utarbeidet randomiseringslisten, hvem rekrutterte pasienter og hvem rekrutterte pasientene
|
|
Blinding
|
11
|
Hvorvidt pasienter, forsøksledere og de som vurderte dataene var blindet for hvilken gruppe pasienten var i, og hvordan blinding ble evaluert etterpå
|
|
Statistiske metoder
|
12
|
Statistiske metoder brukt for å sammenlikne gruppene for primære effektmål, metoder for tilleggsanalyser som subgruppeanalyser og justerte analyser
|
|
Resultater
|
|
|
|
Pasientflyt
|
13
|
Flyt av pasienter gjennom de ulike stadier (diagram er anbefalt). Spesifikt beskrevet antall pasienter rekruttert, randomisert, mottatt intervensjon og analysert for primære effektmål. Beskrivelse av avvik fra opprinnelig protokoll med årsak (frafall pga. bivirkning f.eks.)
|
|
Rekruttering
|
14
|
Datoer for rekruttering og oppfølging av pasienter
|
|
Utgangsverdier
|
15
|
Utgangsverdier – demografiske og kliniske data for hver gruppe
|
|
Antall analysert
|
16
|
Antall pasienter i hver gruppe som ble inkludert i hver analyse, og hvorvidt analysen ble gjort etter behandlingsintensjonsprinsippet. Angi resultatene i absolutte tall hvis hensiktsmessig
|
|
Effektmål og effektstørrelse
|
17
|
Oppsummert resultat for hvert primære og sekundære effektmål og resultatets presisjon (f.eks. 95 % KI)
|
|
Tilleggsanalyser
|
18
|
Oppgi multiplisitetsproblemer ved å angi alle analyser utført, inkludert subgruppeanalyser og justerte analyser, og om de var forhåndsbestemt eller ikke
|
|
Bivirkninger
|
19
|
Alle vesentlige adverse hendelser og bivirkninger i hver gruppe
|
|
Diskusjon
|
|
|
|
Tolking av resultater
|
20
|
Tolking av resultater, i forhold til studiens hypotese, kilder til mulig bias (skjevhet) og fare for multiplisitetsproblemer med analyser og effektmål
|
|
Generaliserbarhet
|
21
|
Generaliserbarhet (ekstern validitet) av hovedfunn i studien
|
|
Studien i større kontekst
|
22
|
Tolking av resultater i sammenheng med tidligere studier av emnet
|
Resultater – hvordan tolke tallene?
Presentasjon av resultatene kan ha stor betydning for hvordan en effektforskjell oppfattes, spesielt dersom man studerer sjeldne hendelser. I tabell 2 viser vi forskjellige måter å angi effekt på. I det ene eksemplet (sinusittstudien) er det en stor andel respondere, og selv om tallene ikke er like, blir inntrykket av behandlingseffekt omtrent det samme enten man viser relative eller absolutte forskjeller i behandlingseffekt. Hvis man derimot betrakter sjeldne hendelser, vil det være av stor betydning for inntrykket om forskjellen er relativ eller absolutt. Et eksempel på dette er reduksjon av risiko for slag hos pasienter med atrieflimmer ved behandling med warfarin eller placebo (9), der den absolutte forskjellen er 3,1 %, mens den relative er 69 %. «Number needed to treat» (NNT) er et mål som er blitt populært blant medisinere. NNT er et uttrykk for hvor mange pasienter som må behandles for at én pasient skal unngå sykdom/død eller oppleve en klinisk relevant effekt. Lave verdier av NNT er altså uttrykk for god effekt av behandling (10). Det er vist at entusiasmen for en og samme behandling avhenger av hvordan resultatet presenteres (11).
I en randomisert studie beskrives resultater som oppnås på gruppenivå. Ved tolking av resultatene er det lett å betrakte pasientene som en homogen gruppe med lik risiko for sykdom, men et av kriteriene for en god studie med høy ekstern validitet er nettopp at den har et bredt spekter av pasienter som har ulik sykdomsintensitet og risiko. Gevinsten ved behandlingen vil være ulik fordi pasientene har ulik utgangsrisiko: Gruppen med høy risiko vil ha større gevinst av behandling (lavere NNT-verdier), mens de med moderat eller liten risiko har mindre gevinst (høyere NNT-verdier). Et eksempel kan være antikoagulasjonsbehandling av pasienter med kronisk atrieflimmer hvor de uten tilleggsrisiko, som tidligere hjerte- og karsykdom, vil ha langt lavere risiko for hjerneslag enn de med tilleggsrisiko. I en artikkel i BMJ fremkom det at spennet i risiko kan være som 1 : 10 i en populasjon med en bestemt tilstand. Det må man ta hensyn til når man evaluerer resultatene og mulig effekt av behandling (12).
Statistisk versus klinisk signifikans
Det er vesentlig å skille mellom begrepene statistisk signifikans og klinisk signifikans (klinisk relevans). Statistisk signifikans forteller ikke noe annet enn at det er mindre enn 5 % sannsynlighet for at den demonstrerte forskjellen mellom to grupper skyldes tilfeldighet og ikke intervensjonen. Klinisk signifikans betyr at den forskjellen som er funnet, også er av klinisk betydning. I ekstremt store forsøk vil man kunne avdekke forskjeller som er så små at de overhodet ikke har klinisk relevans, for eksempel en forskjell i blodtrykk i ulike behandlingsgrupper på 1 – 2 mm Hg. Hva som er klinisk signifikant, vil avhenge av hvilken sykdom det er snakk om og den behandlingen pasienten får. I kreftbehandling vil en 10 % bedring av overlevelse være et stort fremskritt, mens en 10 % endring i varighet av et symptom ved luftveisinfeksjon vil ha liten klinisk betydning.
CONSORT
En rapport fra et randomisert klinisk forsøk skal overbevise leseren om hvorfor studien ble utført og hvordan den ble gjennomført og analysert. For å vurdere en studies sterke og svake sider, må den metodologiske kvaliteten beskrives klart. Det såkalte CONSORT statement (Consolidated Standards of Reporting Trials) (13), ble utviklet for å bedre kvaliteten på artikler som beskriver randomiserte forsøk. Det består av en sjekkliste (tab 3, oversatt til norsk av oss) og et flytdiagram (ikke vist her). I tillegg til at sjekklisten er et godt hjelpemiddel til å skrive en god rapport, kan man også med fordel benytte den i planleggingen av et klinisk forsøk.
Bruk av placebo
Det er diskusjon om når det er riktig å behandle en kontrollgruppe med placebo (14). Hvis det finnes en etablert behandling for en alvorlig sykdom, skal pasientene ifølge Helsinkideklarasjonen sikres den best mulige dokumenterte behandling. Da vil det av de fleste ansees som uetisk å inkludere en gruppe pasienter som kun får placebo. I andre sammenhenger hvor effekten av en medisinsk behandling er usikkert dokumentert, for eksempel vanlige luftveisinfeksjoner i allmennpraksis som oftest er selvbegrensende og ikke livstruende, er det helt nødvendig å sammenlikne med placebo for å vurdere om et medikament har noen effekt i det hele tatt. Dette gjelder de vanligste luftveisinfeksjonene som akutt otitt, akutt sinusitt, akutt bronkitt og akutt tonsilitt. De siste ti årene har det vært gjennomført viktige studier som har endret de vitenskapelige konklusjonene for disse tilstandene. Mens det var svært vanlig å gi antibiotika for alle disse tilstandene i 1980-årene, viser dagens kunnskap at disse tilstandene i stor grad er selvbegrensende, og at mange pasienter ikke har nytte av antibiotikabehandling. Dette hadde det vært umulig å vise uten bruk av placebo.
Diskusjonen om bruk av placebo har vært reist med ny styrke i løpet av de siste par år (15). Bakgrunnen har bl.a. vært studier i den tredje verden med antivirale midler mot HIV-infeksjon. Enkelte har ønsket å starte en slik studie med placebogruppe, men de er blitt kritisert fordi det finnes dokumentert behandling som har effekt.
Forholdet til farmasøytisk industri og etikk er stadig et tema både i pressen og de internasjonale tidsskriftene (16), og debatten er blitt skjerpet vesentlig de siste årene. For de farmasøytiske selskapene er det store interesser som står på spill når det utvikles nye medikamenter, og det kan ikke utelukkes at dette kan påvirke publisering av forskningsresultater. Såkalt publikasjonsbias oppstår fordi såkalte negative studier (der ingen signifikant forskjell mellom behandlinger er vist) sjeldnere blir publisert enn studier som viser effekt av ny behandling (17). Dette gir et skjevt bilde av den reelle effekten av medikamentet. Spørsmålet om publikasjonsbias har for eksempel vært reist ved effekten av de nyere selektive serotoninreopptakshemmerne.
Oppsummerende konklusjoner
-
Kontrollerte kliniske forsøk er velegnet til å finne forskjell mellom grupper av pasienter som utsettes for forskjellige typer intervensjoner.
-
For å kunne trekke pålitelige konklusjoner, er det nødvendig å benytte gode vitenskapelige metoder.
-
Materialet må være tilstrekkelig stort til å kunne trekke holdbare konklusjoner.
-
Intern og ekstern validitet er vesentlig i vurderingen av et klinisk forsøk.
-
Det er vesentlig å skille mellom statistisk signifikans og klinisk signifikans. En statistisk signifikant forskjell trenger ikke bety en vesentlig bedret effekt av behandlingen i den kliniske hverdag.
