Fabrys sykdom ble beskrevet i to uavhengige rapporter av dermatologene William Anderson i England og Johannes Fabry i Tyskland i 1898. I 1967 ble den metabolske defekten kjent som mangel på enzymet ceramidtriheksosidase, som spalter galaktose fra globotriaosylceramid (Gb3) (1). Insidensen av Fabrys sykdom angis fra 1 : 476 000 til 1 : 117 000 levendefødte (2, 3).
Alfagalaktosidasegenet er lokalisert til Xq22, og Fabrys sykdom følger således et X-bundet arvemønster. Det er påvist en rekke ulike mutasjoner i genet, og de fleste familier med forekomst av Fabrys sykdom har sin egen mutasjon. Hemizygote (menn) får vanligvis klassisk Fabrys sykdom, mens mange heterozygote (kvinner) har ganske milde utslag. Rundt 30 % heterozygote utvikler en alvorlig sykdom, og omtrent 80 % av heterozygote kvinner får symptomer på sykdommen (4). Det er derfor ikke riktig å betegne sykdommen som X-bundet recessiv.
Lysosomal akkumulering av glykosfingolipider, hovedsakelig globotriaosylceramid, fører til en multiorgansykdom med affeksjon av hud, nyrer, hjerte og hjerne. I barneårene ser man akroparestesier, teleangiektasier og øyeforandringer, mens nyresvikt, cerebrovaskulær og kardial sykdom tilkommer i voksen alder. Den gjennomsnittlige levealder hos hemizygote er cirka 50 år (5, 6).
Kontrollerte studier de senere år har vist klinisk effekt av intravenøs enzymsubstitusjonsbehandling (7 – 9). Dette er tidligere innført med hell ved Gauchers sykdom (10). Hensikten med denne artikkelen er å omtale to gutter med Fabrys sykdom som er behandlet med enzymsubstitusjon siden november 2001.
Pasient 1. En åtte år gammel gutt fikk tiltakende smerteepisoder i hender og føtter, forsterket ved fysisk aktivitet, og feber av ofte uklar årsak. Etter hvert tilkom magesmerter og diaré. Ti år gammel ble han innlagt ved Barneklinikken, Haukeland Universitetssykehus, høyfebril og med brennende smerter i hender og føtter. Ved klinisk undersøkelse fant man atrofi av muskulaturen i fremre del av foten og en viss atrofi av leggmuskulaturen. Det var ingen hudforandringer. Blodprøver, inkludert revmaprøver og kreatinkinase, var normale. Røntgen og skjelettscintigrafi viste ikke patologiske forhold. Elektromyelografi og nerveledningshastighet gav normale funn. Klinisk mistanke om Fabrys sykdom førte til øyeundersøkelse med påvisning av karakteristiske funn i cornea, conjunctiva, linse og retina. Diagnosen ble bekreftet med meget lave nivåer av enzymet alfagalaktosidase A i leukocytter (0,65 µkat/kg protein, referanseområde 17,7 – 26,4) og høye nivåer av avleiringsproduktet globotriaosylceramid i urinen (160 µmol/mol kreatinin, referanseområde < 10). Genetisk undersøkelse viste en enkeltnukleotiddelesjon i posisjon 10 671 i alfagalaktosidasegenet, hvilket vil føre til leserammeskift og prematur terminering av proteinsyntesen dersom det muterte mRNA-molekylet er stabilt.
Pasient 2. En halvannet år eldre bror hadde hatt liknende, men mindre uttalte symptomer i flere år. Guttenes mor hadde også vært plaget med smerteepisoder som barn. Ved familieutredning fant man lave enzymnivåer og mutasjon i alfagalaktosidasegenet både hos mor (3,0 µkat/kg protein) og hos pasient 2 (2,0 µkat/kg protein). Pasient 2 hadde lærevansker, og nevropsykologisk undersøkelse viste visuell persepsjonsforstyrrelse og nedsatt auditiv hukommelse for sammensatt informasjon.
Behandling. Etter tre år med mye skolefravær og utilfredsstillende symptomkontroll med karbamazepin ble det i november 2001 hos de to guttene, da 13 1/2 og 15 år gamle, startet substitusjonsbehandling med alfagalaktosidase A (Replagal). Behandlingen er blitt gitt som en 40 minutters intravenøs infusjon hver 14. dag.
Behandling
Utredning før oppstart av enzymbehandling viste hos begge hjertestørrelse i øvre grenseområde og PR-intervall i nedre normalområde. Pasient 1 fikk også påvist bradykardi. Cerebral MR viste normale funn hos pasient 1, men små høysignalforandringer i hvit substans hos pasient 2. Det var totalt fem lesjoner, hvorav den største var 6,8 x 4,0 mm (fig 1). Ved utvidet nevrofysiologisk testing fant man hos begge guttene patologisk nerverespons på temperaturendringer, mest uttalt på kulde. Ingen hadde utslett eller mikroalbuminuri.
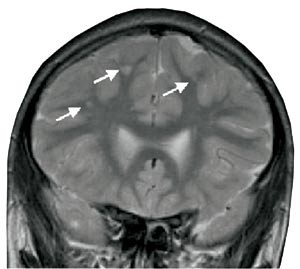
Figur 1 Cerebral MR av pasient 2. Lesjoner før enzymsubstitusjonsbehandling
Etter ett år med enzymsubstitusjon kunne guttene spille fotball og stå på ski uten problemer. De hadde ikke kunnet seponere karbamazepin, men var lite plaget med smerter og hadde ikke lenger diaré. Sommeren 2002 kunne de for første gang på flere år bade i sjøen uten å få smerter. Jevnlige kontroller av globotriaosylceramid i plasma og i urinsediment har vist markant reduksjon av avleiringsproduktet (tab 1). Ekkokardiografi og EKG har ikke vist signifikante endringer, bortsett fra at det hos pasient 1 har utviklet seg en liten mitralinsuffisiens (fig 2). Cerebral MR viser fortsatt normale funn hos pasient 1. Hos pasient 2 er det ingen nytilkomne lesjoner, og T2-signalintensiteten er lett redusert fra første undersøkelse.
|
Tabell 1 Globotriaosylceramid (Gb3)-nivå i plasma og urin før og etter ett års behandling
|
|
Pasient 1
|
|
Pasient 2
|
|
P-Gb3¹
|
U-Gb3²
|
|
P-Gb3
|
U-Gb3
|
|
Før
|
11,3
|
7,4
|
|
9,8
|
21,6
|
|
Etter
|
3,7
|
4,8
|
|
4,1
|
3,7
|
|
[i]
|
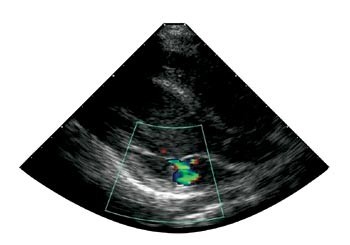
Figur 2 Ekkokardiografi av pasient 1. Mitralinsuffisiens grad 1/4. 2D-ekkokardiografi med fargedoppler fra parasternal langakseposisjon
Diskusjon
Fabrys sykdom er en av flere sjeldne lysosomale avleiringssykdommer. Sykdommen bør regnes å være X-bundet dominant, idet de fleste heterozygote kvinner også får symptomer. Det finnes flere store familier på Vestlandet med Fabrys sykdom. Med utgangspunkt i en av dem ble sykdommen for første gang beskrevet i Norge i Tidsskriftet i 1979 (11). Sykdommen utvikler seg langsomt over år før det oppstår alvorlige symptomer fra nervesystem, hjerte og nyrer. Teleangiektasier er et tidlig symptom og kan lede til diagnose allerede i barndommen. Mørkerøde prikker som ikke lar seg trykke bort og som vanligvis er lokalisert i området mellom lårene og navlen (badebukseområdet), er karakteristiske. De øker i mengde og størrelse over tid (12). Våre pasienter hadde ingen hudforandringer, begge hadde nedsatt svetteevne (hypohidrose) med feberepisoder og plager ved fysisk aktivitet. I likhet med utslett og akroparestesier er hypohidrose en tidlig klinsk manifestasjon.
Flere studier har vist dysfunksjon av temperaturfølsomme afferente nervefibrer. Denne er mer uttalt i føtter enn i hender og mer for kulde enn for varme (13, 14). Ved elektronmikroskopi ser man globotriaosylceramidavleiring og tap av små perifere nervefibrer samt skader på spinalganglienes sensoriske nervefibrer (15). Karbamazepinbehandling hadde effekt på guttenes nevropatiske smerte. Dette førte til økt fysisk aktivitet og redusert muskelatrofi hos pasient 1.
Gastrointestinale symptomer er vanlig forekommende og forårsakes trolig av globotriaosylceramidinnleiring i små kar og ganglier i tarmveggen (vasculopathia og autonom nevropati). De fleste pasienter har luftplager, kvalme, abdominale smerter og diaré (16). Våre pasienter hadde tiltakende mageplager med uttalt, tidvis invalidiserende diaré. Disse plagene forsvant kort tid etter oppstart av enzymsubstitusjonsbehandling.
Øyeforandringer kommer tidlig i sykdomsforløpet. Avleiring i cornea er beskrevet hos et seks måneder gammelt barn (5). Forandringene er karakteristiske med slyngede, utvidede retinale kar og epiteliale avleiringer i cornea (17).
Hjertesykdom debuterer vanligvis i 30 – 40 års alder med konsentrisk hypertrofi av venstre ventrikkel med bevart systolisk funksjon (18). Det er vist at opptil 4 % av menn med hypertrofisk kardiomyopati kan ha Fabrys sykdom (19). Forkortet PR-intervall og abnorm atrioventrikulær overledning samt lett grad av lekkasje over mitral- og aortaklaffen, er også velkjent (20). Begge våre pasienter har moderat hypertrofisk venstre ventrikkel av eksentrisk type og et PR-intervall på 130 ms, som er i nedre normalområdet. I tillegg er pasient 1 relativt bradykard med hvilepuls på 40 – 45 slag/minutt. Ved siste undersøkelse fikk han påvist mitralinsuffisiens grad 1, et funn som kan være oversett ved undersøkelser før enzymsubstitusjonsbehandlingen. Alle disse funnene må imidlertid tolkes som tidlig kardial manifestasjon av Fabrys sykdom.
Ved Fabrys sykdom utvikles over tid cerebrovaskulære lesjoner med varierende symptomer, slik som tinnitus, svimmelhet, hodepine, lærevansker og konsentrasjonsvansker (6). Årsaken kan relateres til progredierende opphopning av globotriaosylceramid i små cerebrale kar som fører til mikroangiopati. Sekundære forandringer ses på T2-vektede MR-snitt som små iskemiske områder i hvit substans og er irreversible. Med stigende alder kan større kar involveres og gi kortikale infarkter av varierende størrelse (21). Funksjonelle cerebrale sirkulasjonsstudier har vist økt vaskulær respons ved Fabrys sykdom som normaliseres ved enzymbehandling (22, 23). Ved slik terapi passerer ikke det gitte enzymet alfagalaktosidase A blod-hjerne-barrieren. De cerebrale forandringene forklares primært av karforandringer, og man håper enzymbehandling vil forebygge cerebrale iskemiske forandringer. Økt trombotisk tendens kan mulig forklares av inflammatoriske forandringer i karveggen (24). Pasient 2 fikk påvist karakteristiske MR-forandringer i hvit substans, hvilket er uvanlig i så ung alder. Den lett reduserte T2-signalintensiteten etter behandlingsstart kan skyldes mindre ødem på grunn av bedret sirkulasjon. Guttens lærevansker og patologiske nevropsykologiske test kan tolkes som sekundære til cerebrale forandringer forårsaket av Fabrys sykdom.
Flere studier har vist effekt av enzymsubstitusjonsbehandling på nyreskader. Man har kunnet vise dels reversjon og dels stans i progrediering av nyresvikten, og tidlig enzymterapi anses å være vesentlig (7 – 9). Våre to pasienter hadde normal kreatininkonsentrasjon og ingen mikroalbuminuri.
Det finnes to tilsynelatende likeverdige enzympreparater på markedet: agalsidase alfa (Replagal), fremstilt i en human cellelinje, og agalsidase beta (Fabrazyme), fremstilt i en ovariecellelinje fra kinesiske hamstere (CHO-celler). Den eneste forskjellen på preparatene er graden og typen av posttranslasjonell glykosylering. Behandlingen tolereres godt, men allergiske reaksjoner kan forekomme. Terapeutisk dose er satt ulikt av de to firmaene, og likeså inklusjonskriteriene i de forskjellige studiene (25). Behandlingen har i flere studier medført nedsatt globotriaosylceramid i plasma og lever-/nyrebiopsier parallelt med bedret nyrefunksjon. Enzymbehandling har også ført til bedret kardial funksjon, bedring av cerebral hyperperfusjon samt lindring av nevropatiske smerter (7, 8, 23).
Praktisk sett kan behandlingen gjennomføres på en sykehuspoliklinikk eller på et legesenter. Hjemmeinfusjon har også vært prøvd. Intravenøs enzymsubstitusjonsbehandling er krevende og kostbart. Pasientene blir etter dagens opplegg avhengige av infusjoner hver annen uke på ubestemt tid. Som alltid når proteiner settes intravenøst, må det overvåkes nøye med henblikk på allergiske reaksjoner. Det behandlingsregime vi per i dag benytter, medfører en årlig medikamentutgift per pasient på 1,5 millioner kroner. Behandlingen må inntil videre vurderes som eksperimentell, og må derfor med jevne mellomrom revurderes både med hensyn til nytteverdi og økonomi. Dersom behandlingsprinsippet innfrir forventningene, vil spørsmålet reise seg om hvem som skal tilbys, eller hvem som vil komme til å kreve, behandling. Skal symptomfrie personer behandles? Hvor tidlig i livet skal behandlingen iverksettes? Skal kvinnelige bærere med lettere symptomer få behandling? Under enhver omstendighet er dette et nytt eksempel på en behandlingsform som er forbeholdt mennesker i den rike del av verden.
