Legemiddeldistribusjonen rundt om i kroppen skjer i all hovedsak med basis i to mekanismer, nemlig diffusjon, som i noen grad er omtalt i en tidligere artikkel om absorpsjon (1), og konveksjon. Diffusjon refererer til den utbredelse molekyler får i et rom på grunn av sin egen bevegelsesenergi, mens konveksjon beskriver forflytting av molekyler med en trykkgradient.
For små molekyler er diffusjonsprosessene viktigst, mens større molekyler fortrinnsvis distribueres ut til vevet med blodbanen ved konveksjon. Diffusjonsprosessene går langs en konsentrasjonsgradient og er ikke spesielt raske eller effektive. Tiden det tar et molekyl å diffundere en viss distanse er proporsjonal med kvadratet av avstanden. Konveksjonsprosessene går raskere. Tidsforbruket til et molekyl som transporteres med konveksjon, er direkte proporsjonalt med avstanden, men forutsetter altså tilstedeværelsen av en trykkgradient.
Fritt og bundet legemiddel
Absorbert legemiddel foreligger i kroppen i en av to tilstander: fritt eller bundet til proteinmolekyler. Det er bare fritt (ubundet) legemiddel som kan krysse biologiske membraner, distribueres rundt i kroppen og elimineres. Det er kun den frie fraksjonen av legemidlet som kan være farmakologisk aktiv og utøve den terapieffekten vi er ute etter. Den bundne fraksjonen er å betrakte som en uvirksom transport- og lagringsform av legemidlet.
Et legemiddels proteinbindingsgrad kvantifiseres vanligvis i plasma og uttrykkes i prosent. Legemidlenes bindingssteder i plasma er avhengig av molekylenes fysikalsk-kjemiske egenskaper. Noen legemiddelgrupper bindes fortrinnsvis til akuttfaseproteiner som for eksempel alfa₁-surt glykoprotein (orosomukoid), mens de fleste plasmabundne legemidler bindes til albumin. Graden av plasmaproteinbinding varierer i betydelig grad mellom ulike legemidler (tab 1) (2). Noen endogene stoffer som også brukes som legemidler, som tyroksin, kortikosteroider og noen kjønnshormoner, er høygradig bundet til spesialiserte bindingsproteiner. Når konsentrasjonen av et legemiddel måles i serum eller i plasma, er det totalkonsentrasjonen, altså konsentrasjonen av fritt pluss bundet legemiddel, som bestemmes.
|
Tabell 1 Proteinbindingsgrad og tilsynelatende distribusjonsvolum for noen velkjente legemidler (2). Det er ingen innbyrdes sammenheng mellom de to variablene; legemidler med høy proteinbindingsgrad kan, som eksemplifisert med digitoksin og warfarin, gjerne ha et lite tilsynelatende distribusjonsvolum
|
|
Virkestoff
|
Proteinbindingsgrad (%)
|
Tilsynelatende distribusjonsvolum (l/kg)
|
|
Amitriptylin
|
95
|
15
|
|
Ciprofloksacin
|
40
|
1,8
|
|
Deksametason
|
68
|
0,8
|
|
Diazepam
|
99
|
1,1
|
|
Digitoksin
|
97
|
0,5
|
|
Digoksin
|
25
|
5,0
|
|
Kloramfenikol
|
53
|
0,9
|
|
Klorokin
|
61
|
115
|
|
Koffein
|
36
|
0,6
|
|
Kokain
|
91
|
2,0
|
|
Litium
|
0
|
0,7
|
|
Tiopental
|
85
|
2,3
|
|
Warfarin
|
99
|
0,1
|
Distribusjonsvolum
Distribusjonsvolumet (egentlig det tilsynelatende distribusjonsvolumet) forteller oss noe om fordelingen av et legemiddel i kroppen. Når distribusjonsvolumet er lite, gjenfinnes mesteparten av legemidlet i plasma. Når distribusjonsvolumet er stort, gjenfinnes mesteparten av legemidlet i vev utenfor plasma. Distribusjonsvolumet uttrykker ikke noe spesifikt anatomisk rom og er på alle måter et abstrakt begrep, men kan likevel gi oss klinisk viktig informasjon om et legemiddels egenskaper.
Et legemiddels distribusjonsvolum er definert som det forholdstallet som multiplisert med legemiddelkonsentrasjonen gir den totale mengden legemiddel i kroppen. Hvis man injiserer et medikament intravenøst og måler plasmakonsentrasjonen av midlet ved noen senere tidspunkter og etter at distribusjonslikevekt har inntrådt, vil man trolig se en eksponentiell reduksjon av plasmanivået, slik som i figur 1. Hvis man projiserer tid-konsentrasjons-forløpet til tiden 0, vil konsentrasjonsaksen krysses ved et punkt som kalles C₀. Denne imaginære og teoretiske størrelsen uttrykker en plasmakonsentrasjon ved en tid 0 og i en situasjon hvor legemidlet er blitt distribuert. Hvis den totale dosen legemiddel som ble inntatt (D), divideres med C₀, får man en proporsjonalitetsfaktor som uttrykker hvordan et legemiddel distribueres i kroppen. Denne faktoren kalles det tilsynelatende distribusjonsvolumet, forkortet Vd eller VD. Formelen skrives vanligvis slik: Vd = D/C₀
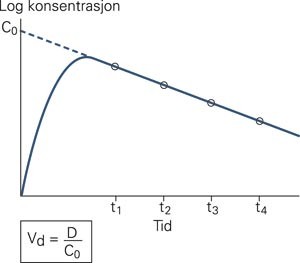
Sammenhengen mellom det tilsynelatende distribusjonsvolumet (Vd), dosen (D) og den tenkte legemiddelkonsentrasjonen ved tiden 0 (C₀). I praksis finner man C₀ ved å ekstrapolere eliminasjonskurven tilbake til tiden 0 i et diagram med logaritmisk skala på y-aksen.
Det tilsynelatende distribusjonsvolumet uttrykkes i enheten liter eller liter/kg. Hvis Vd er lik 1 l/kg, vil legemidlet kunne være jevnt fordelt i hele kroppen. I de tilfellene hvor Vd er større enn 1 l/kg, snakker man om legemidler som fortrinnsvis distribueres ut i vevet, slik at plasmakonsentrasjonen vil være lavere enn vevsnivåene. En rekke stoffer er bundet i utstrakt grad i perifert vev, som for eksempel i lunge-, muskel- og fettvev. Hvis Vd er mindre enn 1 l/kg, vil legemidlet relativt sett finnes distribuert i større grad til plasma enn til annet vev. Er Vd vesentlig mindre enn 0,1 l/kg, betyr det at legemidlene i stor grad kun gjenfinnes i blodbanen, slik det blant annet er for heparin og erytropoietin. Det tilsynelatende distribusjonsvolumet for noen vanlige legemidler er oppgitt i tabell 1 (2).
Proteinbindingsgrad og distribusjonsvolum kan være klinisk viktige i noen situasjoner. For eksempel er det bare legemidler med lav proteinbinding og lite distribusjonsvolum som effektivt kan dialyseres ut av kroppen etter overdoser. Et viktig eksempel på et slikt legemiddel er litium. Motsvarende kan legemidler som er høygradig proteinbundet rundt omkring i kroppens vev og derfor har et stort distribusjonsvolum, diffundere tilbake til blodbanen etter at døden er inntrådt, noe som kan gi opphav til feiltolkninger i rettstoksikologiske sammenhenger.
Spesielle distribusjonshindre
Noen anatomiske strukturer er blitt oppfattet som distribusjonsbarrierer i en slik grad at de fremdeles omtales med termer som blod-hjerne-barrieren og placentabarrieren. Blodkarene i hjernen er omgitt av gliaceller og fremstår som tettere enn andre blodkar både når det gjelder diffusjon og konveksjon, men utgjør ikke noe avgjørende passasjehinder for legemidler. Cerebrospinalvæsken inneholder nesten ikke protein, noe som betyr at likevekten i mengdeforholdet mellom legemiddelnivået på hver av blod-hjerne-barrierens sider blir en funksjon av den frie (ubundne) fraksjonen legemiddel i blod. Dette medfører at legemiddelkonsentrasjonen i cerebrospinalvæsken ofte blir relativt lav i forhold til totalmengden sirkulerende legemiddel. Små, ikke-ioniserte og spesielt svært fettløselige stoffer får vanligvis lett adgang til sentralnervesystemet. Placentabarrieren er i enda mindre grad et passasjehinder. Konseptet om placenta som et filter som slipper gjennom viktige næringsstoffer og hindrer passasje av legemidler, er direkte feilaktig, og placenta bør betraktes som et endokrint organ med viktige transport- og distribusjonsfunksjoner og ikke som en legemiddelbarriere.
For ca. 30 år siden identifiserte forskere en cellulær transmembranpumpe som kunne transportere en rekke forskjellige, kjemisk ubeslektede cytostatika ut av resistente kreftceller mot en stor konsentrasjonsgradient. Denne pumpen, som kalles P-glykoprotein, uttrykkes ikke bare i cellemembranene til resistente kreftceller, men finnes dessuten konstitutivt uttrykt i vev som lever, nyrer, tarmepitel, testikler og ovarier. P-glykoprotein er dessuten uttrykt i kapillarer i blant annet hjernen og placenta. Dette forklarer i noen grad hvorfor cellegifter ikke virker godt ved for eksempel lever- eller nyrekreft, og er dessuten en viktig mekanisme ved ervervet cytostatikaresistens. P-glykoprotein kan ikke bare pumpe cellegifter, men også en rekke andre molekyler (som blant annet inkluderer noen blodtrykksmedisiner, antivirale midler, antibiotika, immunhemmende midler og antidepressiver) ut av cellene, og noen legemidler (kalsiumblokkere, ciklosporin og andre) kan dessuten hemme pumpemekanismen (3). Denne typen interaksjoner har, ved siden av å påvirke fordelingen av legemidler i kroppen, følger for absorpsjon og eliminasjon, og er med på å bestemme responsen på legemiddelbehandling på en mer basal og generell måte enn kun ved noen former for kreftterapi.
Spesielle forhold gjør seg gjeldende også når det gjelder overgang av legemidler til morsmelk. I prinsippet blir legemidler som er svake baser og legemidler som har høy fettløselighet, oppkonsentrert i melken. Det er også en rekke andre forhold som påvirker graden av eksponering hos et barn som ammes. Derfor kan man ikke ut fra syre-base-egenskaper og fettløselighet forutsi hvorvidt det er tilrådelig å amme eller ikke ved bruk av et bestemt legemiddel. I stedet må man søke informasjon om dette i systematiske oversikter som er basert på kliniske studier, som for eksempel (4).
