Vogt-Koyanagi-Haradas sykdom er en sjelden form for bilateral panuveitt. Sykdommen ble opprinnelig oppfattet som flere ulike tilstander, beskrevet av henholdsvis Vogt i 1906, Koyanagi i 1929 og Harada i 1926. Disse tilfellene er senere ansett som varierende utrykk for samme tilstand.
Sykdommen debuterer vanligvis i ung voksen alder, men forekommer i alle aldersgrupper. Den er hyppigst i Asia og Sør-Amerika. Det er ofte prodromalsymptomer i form av hodepine, tinnitus og kvalme før øyet affiseres. Deretter ses bilateral, vanligvis simultan uveitt med rask synsreduksjon. Forløpet er langvarig, med depigmentering av øyebunnen og varig synsnedsettelse. Tilstanden responderer vanligvis godt på systemiske steroider, men residiverende inflammasjon ses i en del tilfeller. Vi presenterer her tre kasuistikker som illustrerer sykdomsbildet.
Pasient 1. En 29 år gammel tidligere frisk mann, opprinnelig fra Colombia, ble innlagt i september 1999 grunnet synsreduksjon ledsaget av hodepine i en uke. Begge øyne var injiserte, med presipitater på corneas bakflate samt moderat korpuskulær lysvei i forkammeret og i corpus vitreum. Det ble også påvist papillødem, koroidal effusjon og subretinalvæske. Visus var 0,16 på høyre øye og 0,125 på venstre. Fluoresceinangiografi viste multifokal hyperfluorescens, svarende til pigmentepitellaget og lekkasje subretinalt. Ultralydundersøkelse verifiserte koroidal effusjon og eksudativ netthinneløsning (fig 1a). Vanlige blodprøver var normale. Serologiske prøver gav ikke holdepunkter for tilstedeværelse av aktuell infeksjon. Waalers prøve, ANA og HLA-B27 var negative. I spinalvæsken var antall leukocytter forhøyet (182 · 10⁶/l). Cerebral MR viste væske i mellomøret, ellers var det normale funn. Røntgen thorax var normalt. Nevrolog, øre-nese-hals-lege og hudlege fant normale forhold.
Pasienten ble behandlet med prednisolon 100 mg daglig i avtrappende dosering samt deksametason øyedråper, hvilket førte til avtakende inflammasjon og tilbakegang av eksudasjonen i øyebunnen. Ved kontroll fire måneder etter innleggelsen var visus 0,3 på høyre øye og 0,25 på venstre. Det var da ingen tegn til subretinalvæske, men karakteristiske fundusforandringer med flekkevis depigmentering og kornet pigmentering (fig 1b). Pasienten er senere fulgt opp ved annet sykehus.
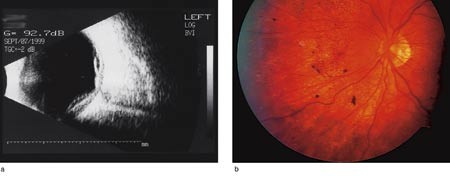
Figur 1 Pasient 1, venstre øye. a) Ultralydundersøkelse viser koroidal effusjon og serøs netthinneløsning. b) Flekkevis depigmentering og kornet pigmentering i stadium 3
Pasient 2. En 50 år gammel tidligere øyefrisk kvinne, opprinnelig fra Filippinene, ble innlagt til utredning i desember 2001. Visus ved innleggelse var 0,7 på høyre øye og 0,3 på venstre. Begge øyne var lett injiserte, med en svak korpuskulær lysvei i fremre og bakre segment, ødem i macula og serøs netthinneløsning bilateralt. Fluoresceinangiografi viste multifokal hyperfluorescens i pigmentepitellaget, med gradvis økende lekkasje subretinalt. Ultralydundersøkelse viste serøs netthinneløsning i begge øyne. Vanlige blodprøver viste SR 72 mm, kreatinin 144 – 97 µmol/l, ellers normalt. Hun var HLA-B27- og ANA-positiv. Pasienten ønsket ikke spinalpunksjon, men MR av hodet viste kontrastoppladning i choroidea. Røntgen thorax var normalt. Øre-nese-hals-lege, hudlege og nevrolog påviste intet patologisk.
Pasienten fikk prednisolon 80 mg daglig i nedtrappende dosering, med god effekt. Ved utskrivning etter vel en uke var subretinalvæsken nærmest fullstendig resorbert, og visus var 1,5 på høyre øye og 1,0 på venstre. I 2003 ble det påvist netthinneforandringer relatert til annen sykdom, men ved senere kontroller har det ikke vært tegn til uveitt eller koroidal effusjon. Ved kontroll 28 måneder etter sykdomsdebut var visus 0,8 på høyre øye og 1,0 på venstre.
Pasient 3. En 13 år gammel tidligere frisk gutt, med far fra Peru og norsk mor, ble innlagt november 2002. Han hadde hatt gradvis økende hodepine i to uker, og fra innleggelsesdagen redusert syn og metamorfopsier på høyre øye. Visus var 0,4 på høyre øye og 1,2 på venstre. Det var ikke tegn til intraokulær inflammasjon, bortsett fra en stor, avgrenset lesjon temporalt i høyre fundus, omgitt av subretinalt ødem og serøs netthinneløsning nedad (fig 2a). Utredning med CT og MR cerebrum og orbita, røntgen thorax og ultralyd abdomen var alle normale. Fundusforandringene i høyre øye progredierte raskt, og fire dager etter innleggelsen var det blitt tilsvarende forandringer også i det venstre. Fluoresceinangiografi viste hyperfluorescens med multifokal lekkasje (fig 2b). Pediater, øre-nese-hals-lege og hudlege fant normale forhold. I spinalvæsken var det normalt celletall. Vanlige blodprøver var normale. Serologiske prøver gav ikke holdepunkter for tilstedeværelse av aktuell infeksjon.
Pasienten fikk prednisolon 40 mg daglig, med rask effekt. Medisindosen ble gradvis redusert i løpet av de påfølgende 12 måneder, og behandlingen førte til en fullstendig tilbakegang av eksudasjonen og normalisering av visus på begge øyne. En viss grad av depigmentering var synlig i områder med tidligere eksudasjon, men ellers var det ikke tegn til andre komplikasjoner. Ved kontroll 18 måneder etter sykdomsdebut var visus 1,5 på begge øyne, og pasienten var symptomfri.

Figur 2 Pasient 3, høyre øye. a) Avgrenset lesjon med koroidal inflammasjon og subretinalt ødem temporalt for macula. b) Fluoresceinangiografi viser hyperfluorescens med multifokal lekkasje
Diskusjon
Vogt-Koyanagi-Haradas sykdom er en kronisk, bilateral granulomatøs panuveitt som ofte er assosiert med affeksjon av sentralnervesystemet, mellomøret og huden. Det kliniske bildet gir mange differensialdiagnostiske muligheter (ramme 1). Årsaken er ukjent, men T-cellemediert autoimmunitet rettet mot melanocytter i de involverte organer er beskrevet (1). Antistoffer mot fotoreseptorer og Müller-celler er også påvist i serum. Om sykdommen forårsakes av en utløsende faktor, som for eksempel en virusinfeksjon, har vært diskutert, men dette har man foreløpig ikke kunnet verifisere. Enkelte HLA-typer er assosiert med tilstanden, særlig HLA-DR4, HLA-Drw53 og HLA-DQw3. Tilstanden forekommer oftest i befolkningsgrupper med økt pigmentering, slik som hos alle våre pasienter. I Asia, India, Sør-Amerika og Midtøsten er sykdommen relativt hyppig. Prevalensen er i Japan angitt til 9,2 % av uveittpasienter (2). I USA og Nord-Europa er tilstanden sjelden (3, 4).
Ramme 1
Differensialdiagnoser ved Vogt-Koyanagi-Haradas sykdom
Vogt-Koyanagi-Haradas sykdom inndeles i fire stadier. Stadium 1 (prodromalstadium) inntreffer dager eller få uker før øyesymptomene. Typisk er hodepine, smerter i orbita, kvalme, lett feber, meningisme og tinnitus. Stadium 2 (uveittstadium) varer fra uker til måneder. Pasienten utvikler gradvis en bilateral uveitt. Den koroidale inflammasjonen gir papillødem, eksudativ netthinneløsning og pigmentepitelaffeksjon med betydelig synsnedsettelse. Stadium 3 (konvalesens/kronisk stadium) fører til at uveitten avtar, samtidig som det er økende depigmentering av fundus, i noen tilfeller også subretinal fibrosering og koroidal neovaskularisering. Poliose, vitiligo og alopesi kan forekomme. Stadium 4 (tilbakefallsstadium) preges av fremre uveitt og komplikasjoner i form av bakre synekier, katarakt og glaukom. I vårt materiale hadde pasient 1 og pasient 3 begge typiske prodromalsymptomer. Pasient 1 var den som hadde mest uttalt øyeaffeksjon, men også kortest oppfølgingstid i vår avdeling, slik at eventuell depigmentering av hud i stadium 3 er ukjent. Pasient 2 og pasient 3 utviklet heller ikke hudaffeksjon. Diagnostiske kriterier er foreslått av American Uveitis Society og senere modifisert. Såkalt komplett sykdom innbefatter affeksjon av både øye, hud og sentralnervesystem. Ved inkomplett sykdom må enten hud eller sentralnervesystem være affisert, og ved sannsynlig Vogt-Koyanagi-Haradas sykdom er ren øyeaffeksjon tilstrekkelig, dog med samme omfattende kriterier til øyeaffeksjonens art (5).
Det kliniske forløpet varierer – fra en selvbegrensende inflammasjon til alvorlig kronisk eller residiverende inflammasjon med beskjeden behandlingsrespons og høy komplikasjonsfrekvens (6). Ubehandlet er prognosen dårlig, og tidlig oppstart med systemiske kortikosteroider er vesentlig for å modifisere sykdomsforløpet og redusere komplikasjonsrisikoen. 50 – 70 % av pasientene oppnår visus bedre enn 0,4 etter adekvat steroidbehandling (6, 7). I enkelte studier er det anført en dårligere prognose hos unge pasienter (8). Stadium 4 er assosiert med betydelig visusreduksjon og høy komplikasjonsrisiko. Kronisk inflammasjon (mer enn seks måneder) forekommer hos 17 – 40 % av pasientene, på tross av adekvat behandling. Depigmentering og karakteristiske forandringer i fundus finnes hos over 90 % av disse, mot hos omkring 60 % av pasienter uten langvarig inflammasjon (9). Ved tidlig, adekvat behandling reduseres risikoen for kronisk inflammasjon og depigmentering (6). Read og medarbeidere fant at blant 101 pasienter utviklet 53 % minst én komplikasjon (6). De vanligste komplikasjonene var katarakt (44 %) og glaukom (29 %). Mindre vanlig var koroidal neovaskularisering (15 %) og subretinal fibrosering (8 %). Disse pasientene hadde et dårligere visusmessig resultat, spesielt ved utvikling av mer enn én komplikasjon.
Vårt pasientmateriale illustrerer sykdomsbildet og det kliniske forløpet ved Vogt-Koyanagi-Haradas sykdom. Pasientene ble alle undersøkt og utredet forholdsvis kort tid etter symptomdebut, og de fikk rask og adekvat behandling. Sykdommen forekommer sjelden i Norge, men diagnosen må has in mente hos innvandrere og pasienter med utenlandsk opprinnelse. Tidlig diagnose og rask behandling med systemiske steroider reduserer faren for varig synsreduksjon.
