Patients with heart failure often use many drugs, of which several are metabolised via cytochrome P-450 (CYP) enzymes. This is a group of enzymes where genetic polymorphism may lead to large individual differences in metabolism.
A 60-year-old man born in Kashmir was admitted to Ullevaal University Hospital with an ST- elevation myocardial infarction (STEMI). He had a history of known diabetes Type 2 and hypercholesterolaemia. Time from symptom onset to coronary angiography was 150 min. An occluded left anterior descending artery (LAD) was identified and successfully stented. The patient developed post-infarction heart failure with a reduced function of the left ventricle, an almost akinetic anterior wall and an ejection fraction of 30 to 35. After initial treatment he was discharged with the following drugs: simvastatin 40 mg daily, metoprolol 25 mg daily, ramipril 5 mg daily, clopidogrel 75 mg daily, acetylsalicylic acid 75 mg daily and glimepiride 4 mg daily. He was later followed up at the Heart failure clinic at Lovisenberg Deaconal Hospital (Oslo) where he complained of a bothersome dry cough during a control a month after hospitalisation. This was associated with ramipril, which was discontinued and replaced with losartan 50 mg daily.
Dry cough is an adverse event that affects about 10 of patients treated with ramipril and other ACE inhibitors (1). The mechanism behind dry cough is accumulation of bradykinin, a substance which is also metabolised by angiotensin I converting enzyme (ACE). ACE inhibitors reduce inactivation of bradykinin and activation of angiotensin I to angiotensin II.
Angiotensin II receptor antagonists work directly by blocking receptor binding of angiotensin II and do not interfere with metabolism of bradykinin. This is why angiotensin II receptor antagonists are not associated with the adverse event dry cough. Angiotensin II inhibitors are indicated for treatment of patients with heart failure and intolerance to ACE inhibitor. Losartan was chosen as an alternative to ramipril in this case, but there are also other angiotensin II receptor antagonists that have the indication heart failure.
At a follow-up five weeks after conversion to losartan, an exercise-test indicated deterioration of the heart failure. The test could not be completed because the patient developed dyspnea and exhaustion. Eccocardiography showed fluid in the right pleura and a thrombus in the left ventricle, but there was no measurable reduction in the ejection fraction. Chest X-ray showed the same failure pattern with increasing basolateral fluid collection on the right side. The patient was hospitalised to start warfarin treatment and for intravenous dehydration with furosemide. Dosing with warfarin was started according to a standard protocol with three tablets (7.5 mg) the first and second day. On Day 2 of the treatment the INR value was 3.1 (therapeutic window 2.0 – 3 – 0). The INR value continued to be above the therapeutic window and on Day 9 (without a new dose) it was measured to be 4.9 (fig 1). At the next measurement, Day 13, the INR value had decreased to 4.0, and a new warfarin dose of two tablets was first given four days later.
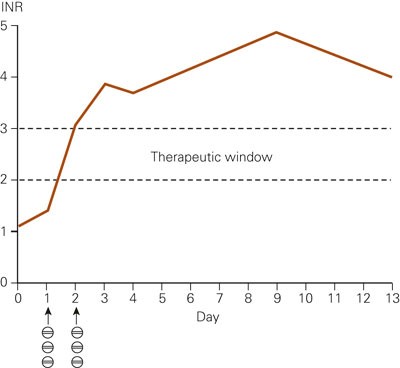
Figure 1 INR measurements after start of warfarin treatment (three tablets/7.5mg on Day 1 and Day 2)
Even if there was no measurable change in the ejection fraction, clinical symptoms and examinations showed clear signs of worsening of the heart failure. The worsening was not associated with a change from ramipril to losartan, but was assessed as a natural development of the disease. A well-known clinical phenomenon with warfarin is a large individual variation in dosing needed to achieve a therapeutic INR value. However, it is rare that patients get an elevated INR value that lasts for more than ten days after the first two doses. The maximal INR level is unknown, but the patient’s INR value decreased from 4.9 to 4.0 between Day 9 and Day 13 of the warfarin treatment. The persistent high INR values may indicate a substantially longer half-life for warfarin than normal (1 – 2 days (2)). This has been described for warfarin in patients with mutations in the gene that codes for the enzyme CYP2C9 (3).
A persistent high INR value after the start dose raised the suspicion that the patient had a genetically based slow metabolism of warfarin. To evaluate if this was the mechanism behind the high INR value, a pharmacogenetic analysis of cytochrome P-450 (CYP) mutations was requested. A blood sample was sent to Department of Psychopharmacology at Diakonhjemmet Hospital (Oslo) for analysis. The analysis showed that the patient was a homozygous carrier of mutations in both CYP2C9 and CYP2D6; i.e. mutation 1075A>C (allele CYP2C9*3) and mutation 1846G>A (allele CYP2D6*4). Before the test result was available, the patient had again been hospitalised because of dyspnoea, chest pain and pulmonary congestion.
Frequent CYP mutations have not been described for the population in Kashmir, but among white people, CYP2C9*3 has an allele frequency of about 7 and CYP2D6*4 a frequency of about 20 (4). CYP2C9*3 and CYP2D6*4 code for a markedly lower metabolism activity (< 5 of the normal) than the wild-type. The patient was homozygous for both alleles, and therefore was a slow metaboliser both via CYP2C9*3 and CYP2D6*4. Slow metabolism via CYP2C9 explained the persistent high INR value after the start dose of warfarin. Added to the test result was a note that the clinical effects of losartan and glimepiride would be affected by a slow CYP2C9 metabolism, whereas the level of metoprol would be affected by a slow CYP2D6 metabolism. For losartan it has been claimed that a reduced CYP2C9 metabolism probably means a missing angiotensin II blockade, as the substance is metabolised into a 10 to 40 times more active substance via CYP2C9 (5). It was therefore recommended that losartan was replaced with an alternative angiotensin II receptor antagonist.
Based on the answer from the pharmacogenetic CYP analysis, the worsening of the heart failure after changing from ACE inhibitor to losartan was associated with the patient’s missing ability to create an active metabolite from losartan. It was therefore decided to substitute losartan with an angiotensin II receptor antagonist that was not metabolised by CYP2C9. The choice was valsartan that was titrated up from 40 mg daily to the target dose of 160 mg daily.
The patient is still followed up at the heart failure clinic. There has not been any new admissions or events with dyspnea or pulmonal congestion after changing the angiotensin II receptor antagonist. After three months the ejection fraction had improved to about 45. The patient felt less short-breathed than earlier and was able to walk up two to three floors without stopping. Blood pressure was about 110/75, heart beat 70 and fasting blood sugar 6 mmol/L. He was still treated with warfarin, and the INR value was stable in the area 2 – 2.5 with a weekly dose of 1.5 – 2.5 tablets.
Discussion
Enzymes within the CYP family are important for the metabolism of many drugs. These enzymes show large individual differences in phenotype. This may be related to genetic conditions, diseases and intake of enzyme inhibitors or inducers. Genetic differences are typical for CYP2C9 and CYP2D6, where inherited mutations usually lead to a reduced or defect metabolism (6).
The patient turned out to be a homozygous carrier of mutations that code for slow metabolism via CYP2C9 and CYP2D6. Four of the prescribed drugs are metabolised extensively by CYP2C9 (warfarin, losartan and glimepiride) or CYP2D6 (metoprolol) (4). Losartan is a prodrug and is activated by the enzyme in question, while the three others are administered in a pharmacologically active form and become inactivated (fig 2). Clinically this means that the patient had a reduced formation of active losartan metabolite (probably less than 10 of the normal) and thereby insufficient angiotensin II blockade (5). For warfarin, glimepiride and metoprolol a slow metabolism means a high concentration in relation to the administered dose (about five to ten times higher than normal) and thereby an increased risk of adverse events (4). Warfarin is a drug with a narrow therapeutic window and increased bleeding risk with elevated INR values, but no sign of bleeding was observed in the patient.
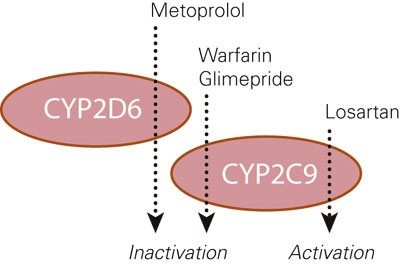
Figure 2 Schematic presentation of the CYP metabolism that was strongly reduced in our patient
Insufficient angiotensin II blockade of losartan because of slow metabolism coincided with a deterioration of the heart failure that was observed at control five weeks after changing from the ACE inhibitor ramipril. At the same control a thrombus was observed in the left ventricle. It cannot be excluded that worsening of the heart failure was a result of the thrombus, but we consider lack of angiotensin II inhibition after the switch to losartan to be a more probable cause. In that case one may speculate whether the lack of an effect from losartan also caused the thrombus and the need for treatment with warfarin.
The reason for requesting the pharmacogenetic CYP analysis was the persistently high INR value after the starting doses of warfarin. The test result confirmed that a high INR value was caused by a genetic slow metabolism of warfarin and not by other factors such as lack of compliance or interaction with other drugs, foods or herbal medicines. The clarification probably contributed to alleviating a stressful situation, not least for the patient. It also gave further grounds for treatment with warfarin requiring that special precaution is taken with dose changes, other treatment and food. In addition, the test answer increased our understanding of the other drugs that the patients were treated with. Losartan was replaced by another angiotensin II receptor antagonist, while a metoprolol dose low enough to be tolerated seemed like a rational choice.
CYP mutations are stable throughout life. Information from pharmacogenetic analyses therefore has a prospective value for choice of possible drug treatment. Comorbidity with heart failure is common and it is reasonable to assume that the information may become useful for our patient later. However, there are challenges associated with the flow of information and knowledge about clinical use of results from pharmacogenetic analyses in the health services. Out-patient follow-up may contribute to solving these challenges through continuous assessments of health status and drug use.
The Journal of the Norwegian Medical Association No. 18/2006 had pharmacogenetics as a theme and focused on individual adaptation of treatment as a vision for the future. CYP genotyping is currently a routine analysis in several Norwegian laboratories and a tool for rationalizing choice of drug and dose to each patient. However, the cost-benefit of CYP genotyping is debated and a core question is to what extent the tests should be used. Questions that concern health priorities are complicated and difficult, but it is interesting that some hospitals in Denmark have established routine CYP genotyping of all admitted patients (7, 8). This is done from the perspective that investment in this one-time test will be cost-effective in the long run in the form of fewer adverse events, less need for follow-up, better treatment effect and fewer admissions. A prerequisite for such analysis to be useful is the existence of concrete recommendations on choice of drug and start dose for patients with different CYP genotypes (9).
CYP genotyping consists of about ten separate mutation analyses with a total cost that corresponds to that for three months use of losartan (based on today’s costs). In our case, the treatment with losartan was probably ineffective and may have caused extra admissions and treatment needs. This illustrates that a one-time investment in CYP genotyping may be economical and health- promoting in some patients, but that there is still a need for research to evaluate the usefulness in a larger scale (10).
