I 1882 beskrev den franske hudlegen Philippe Gaucher en pasient med utpreget hepatosplenomegali (1). Han trodde opprinnelig det dreide seg om en godartet leukemivariant. Epstein fant i 1924 at pasienter med denne lidelsen hadde intracellulær lipidopphopning (2). Autosomalt recessiv arvegang ble påvist i 1948 (3), og i 1965 beskrev Brady og medarbeidere enzymdefekten som forårsaker Gauchers sykdom (4). Det ble kartlagt at tilstanden forårsakes av lysosomal akkumulering av sfingolipidet glukosylceramid som følge av redusert aktivitet av hydrolasen glukocerebrosidase. Dette kan særlig føre til svekket beinmargsfunksjon, skjelettplager og påvirkning av sentralnervesystemet. Vi presenterer her to sykehistorier for å illustrere særskilte trekk ved denne sykdommen.
Pasient 1. En ugift, barnløs kvinne født i 1963 ble som tiåring henvist til sykehus fordi hun i flere år hadde vært plaget av neseblødninger. Det var ingen opphopning av familiære sykdommer, spesielt ikke blødningstilstander. Ved innkomst var Hb 8,4 g/100 ml, trombocytter 127 · 10⁹/l og leukocyttallet 5,0 · 10⁹/l med normal differensialfordeling. Klinisk tilstand var upåfallende, bortsett fra en forstørret milt. Mistanken om leukemi ble avkreftet ved beinmargsbiopsi, som derimot viste spesielle celler som ved Gauchers sykdom. Senere ble diagnosen verifisert med måling av glukosylceramid i erytrocyttene: 9,6
I 1997 ble hun igjen undersøkt i sykehus pga. tiltakende menoragi. Da hadde trombocyttallet sunket til 87 · 10⁹/l, Hb var 10,6 g/100 ml, mens leukocyttallet fortsatt var normalt (5,0 · 10⁹/l). Hun hadde normale verdier for elektrolytter, nyre- og leverfunksjon. Røntgenundersøkelser viste begynnende affeksjon av lårbeina.
Man fant indikasjon for enzymsubstitusjonsbehandling. Pasienten setter nå selv intravenøse injeksjoner med rekombinant
Pasient 2. En 35 år gammel gift, barnløs kvinne, askenas, begynte for et par år siden å få tiltakende hoftesmerter. Hun var da bosatt utenlands og tok selv initiativet til bestemmelse av glukocerebrosidaseaktivitet samt genotyping med tanke på Gauchers sykdom. Resultatene av disse undersøkelsene viste at hun hadde lav enzymaktivitet – 1,1 nmol/mg protein per time (nedre referansegrense er 13,0 nmol/mg protein per time) – og at hun var homozygot for en kjent Gaucher-relatert mutasjon i
Siste målinger viste Hb 12,2 g /100 ml, trombocytter 64 · 10⁹/l, leukocytter 7,2 · 10⁹/l og normal differensialfordeling. Hun har ikke blødningstendens. Verken milt eller lever er forstørret. MR av bekken og hofter har ikke vist sikre patologiske funn. Klinisk er hun kjekk og i full jobb. Foreløpig får hun ingen behandling.
Diskusjon
Det er vanlig å dele Gauchers sykdom inn i tre hovedgrupper basert på alder, organaffeksjon og forventet levetid (tab 1) (5 – 7). Kvinner og menn affiseres tilsynelatende likt. Fordi sykdommen er sjelden, er forekomsten usikker. Antall pasienter i Norge med Gauchers sykdom er ikke kjent, men på bakgrunn av opplysninger fra andre europeiske land kan det anslås til 30 – 40 personer, der de aller fleste mest sannsynlig har type 1. Fem av disse får enzymsubstitusjonsbehandling (H. Hauerholt, personlig meddelelse).
Den lysosomale akkumuleringen av glukosylceramid som følge av nedsatt glukocerebrosidaseaktivitet er særlig fremtredende i monocytter/makrofager i det retikuloendoteliale system (lever, milt og beinmarg). Disse såkalte gauchercellene er morfologisk karakteristiske og vanligvis lette å se i beinmargsutstryk (fig 1), men liknende celler kan også sees ved andre lipidavleiringstilstander og ved maligne blodsykdommer (8). Det er rimelig å tro at infiltrasjon i beinmargen med slike fettrike celler etter hvert vil fortrenge den normale hematopoese og dermed føre til symptomer på beinmargssvikt, i første rekke blødningstendens, men også anemi og økt infeksjonstilbøyelighet. Pancytopeni tilskrives også forstørret milt med økt nedbrytning av erytrocytter og trombocytter (hypersplenisme). Debutsymptomene hos vår første pasient var nettopp anemi og blødningstendens til tross for normal trombocyttkonsentrasjon. Kanskje avspeiler dette en redusert trombocyttfunksjon som følge av glukosylceramidakkumulering i beinmargens celler.
I tillegg til beinmargsaffeksjon er skjelettplager vanlig. En rekke beinsykdommer er assosiert med Gauchers sykdom, blant annet osteoporose, avaskulær nekrose (f.eks. Calvé-Legg-Perthes sykdom) og destruksjon av leddflater (9, 10). Radiologisk regnes deformiteter av lårbeina (erlenmeyerkolbedeformitet) som karakteristisk for Gauchers sykdom (fig 2), noe begge våre pasienter fremviste. Denne deformiteten skyldes nedsatt evne til remodellering av knoklene. Ubehandlet kan skjelettforandringene bli så uttalte at de medfører spontanfrakturer og alvorlig invaliditet.
Ulike manifestasjoner fra sentralnervesystemet utgjør den tredje hovedgruppen av plager relatert til Gauchers sykdom, og sees hos pasienter med sykdom av type 2 og type 3. De vanligste er øyeaffeksjon (skjeling), spasmer og mental retardasjon. Vekst og motorisk utvikling hos barn med Gauchers sykdom kan også være redusert (5, 6).
Sjeldnere utvikler pasientene tegn på nedsatt funksjon i respirasjons- og sirkulasjonsorganene (5, 6, 8). Det er angitt at pasienter med Gauchers sykdom er disponert for utvikling av visse neoplasier, blant annet maligne blodsykdommer og beinsvulster (11, 12). Dette er imidlertid ikke endelig klarlagt. Det er antatt at verken kvinnelig eller mannlig fertilitet er affisert, men gynekologiske og obstetriske komplikasjoner utgjør en tilleggsbyrde hos kvinner med Gauchers sykdom (13, 14).
|
Tabell 1 Inndeling av Gauchers sykdom basert på (5 – 7)
|
|
Type 1
|
Type 2
|
Type 3
|
|
Parameter
|
Ikke-sentralnervøs form
|
Akutt sentralnervøs form
|
Juvenil, sentralnervøs form
|
|
Aldersgruppe
|
Unge, voksne
|
Spedbarn
|
Barn, unge
|
|
|
|
|
|
Affisert organ
|
Bein, beinmarg, milt og lever
|
Sentralnervesystemet, milt og lever
|
Sentralnervesystemet, bein, beinmarg, milt og lever
|
|
|
|
|
|
Progrediering
|
Svært variabel
|
Hurtig
|
Variabel
|
|
|
|
|
|
Levealder
|
Normal til noe forkortet
|
Død før 2-års alder
|
20 – 30 år
|
|
|
|
|
|
Insidens¹
|
1/40 000 – 1/200 000
|
< 1/100 000
|
< 1/50 000
|
|
[i]
|
Diagnostikk
Det finnes ingen formelle kriterier for diagnostisering av Gauchers sykdom. Utredning settes oftest i gang på grunnlag av klinisk mistanke, familiær forekomst og eventuell spesiell etnisk tilhørighet, slik pasient 2 demonstrerer. Undersøkelse av beinmarg med påvisning av gaucherceller samt radiologiske funn av skjelettmanifestasjoner styrker mistanken.
Direkte måling av glukocerebrosidaseaktivitet i leukocytter fra fullblod er nødvendig for å kunne stille en sikker diagnose. Det er imidlertid ingen direkte sammenheng mellom grad av redusert enzymaktivitet i leukocytter og kliniske symptomer. Oftest genotypes pasientenes leukocytter også. Det er påvist en rekke ulike mutasjoner. Ved type 1, som omfatter omtrent 90 % av alle med Gauchers sykdom, er punktmutasjonen N370S (utbytting av aminosyren asparagin med serin i posisjon 370 i proteinet) den hyppigste (15). Den fant vi i begge våre pasienter, som har type 1-formen. Nevrologiske symptomer er meget sjeldent hos pasienter med denne mutasjonen. Andre mutasjoner er vanlige ved Gauchers sykdom type 2 (16). Pasienter med samme mutasjon kan ha ulike plager, slik at korrelasjonen mellom feno- og genotype ofte er svak. Imidlertid er det kjent at visse mutasjoner i sterkere grad disponerer for bestemte organmanifestasjoner. For eksempel er pasienter homozygote for punktmutasjonen D409H (utbytting av aminosyren asparginsyre med histidin i posisjon 409 i proteinet) utsatt for hjerteklaffefeil som ofte krever kirurgisk korreksjon i ung alder (17). Verken enzymmåling eller genotyping utføres i Norge, men slike undersøkelser utføres ved ulike laboratorier i Europa, bl.a. i Sverige og Nederland.
Det er gjort en rekke undersøkelser for å se om forskjellige markører også kan være til hjelp ved diagnostisering og oppfølging av behandling ved Gauchers sykdom, blant annet ferritin og en rekke enzymer som alkalisk fosfatase og angiotensinkonverterende enzym (18). Den mest lovende er kanskje chitotriosidaseaktiviteten i plasma (19). Den var klart forhøyet hos begge våre pasienter. Ingen av disse markørene er absolutt spesifikke for Gauchers sykdom.
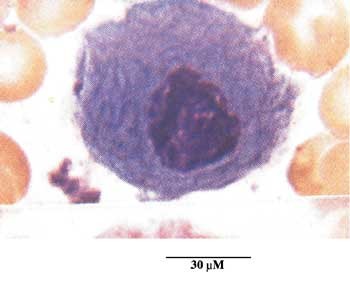
Figur 1 Makrofag med akkumulering av sfingolipider (gauchercelle). Snittet er hentet fra et beinmargsaspirat og farget med hematoksylin/eosin

Figur 2 Røntgenologisk bilde av distale del av a) et normalt lårbein og b) et med såkalt erlenmeyerkolbedeformitet
Behandling
Før man klarte å renfremstille glukocerebrosidaseenzymet, hadde man ingen spesifikk terapi mot sykdommen. Oftest avventet man dersom plagene ikke var for uttalte. Ved alvorlige tilfeller kunne man ty til blodtransfusjon, splenektomi, ortopedisk kirurgi samt behandling med analgetika. Vår første pasient, som fikk diagnosen allerede for 29 år siden, fikk ingen spesifikk behandling de første 25 årene.
Den meste anvendte terapi i dag består i intravenøse infusjoner med glukocerebrosidase. Legemidlet leveres som et pulver som rekonstitueres med sterilt vann og fortynnes i 0,9 % NaCl før det infunderes over 1 – 2 timer, vanligvis én gang annenhver uke. Man kan undre segover hvordan et enzym gitt intravenøst i intakt form finner veien til det indre av de cellene der det skal utøve sin virkning. I sin native form opptas ikke glukocerebrosidase i makrofagene som lagrer glukosylceramid. Det viste seg imidlertid at nedbrytning av en oligosakkarid sidekjede på enzymet slik at mannose eksponeres, fører til binding til en mannosespesifikk reseptor på makrofagmembranen og opptak via endocytose. Intravenøs tilførsel avenzymet synes ikke å medføre noen særlig bivirkninger, og det antas ikke å være kontraindisert ved svangerskap. Erfaringen med denne behandlingen viser god effekt på organomegali, anemi og trombocytopeni (20, 21). Derimot er nytten vedrørende skjelettmanifestasjonene noe mer usikker (10, 22). Det foreligger ikke overbevisende dokumentasjon på at enzymterapi har effekt på sykdom i nervesystemet (20).
Enzymtilskudd er særlig aktuelt for pasienter med type 1-formen. Substitusjonsbehandling med det manglende enzymet har vært tilgjengelig fra 1990. Først benyttet man enzym renset fra placenta, men nå er det tilgjengelig i rekombinant form som imiglukerase. Det er dokumentert at behandlingen reduserer miltstørrelsen, forbedrer hematologiske verdier og biokjemiske indikatorer, reduserer skjelettsmerter og gir kompensatorisk vekstøkning hos barn (13). Det er vist at pasientene får en klart bedret livskvalitet ved enzymbehandling (23). Det er imidlertid store kostnader forbundet med dette (20).
Optimal dosering er ikke klarlagt. Det er sannsynlig at dosen kan reduseres ved tilfredsstillende behandlingsrespons. Hvorvidt man også kan ta behandlingspauser, er omdiskutert (24). Dette har ført til at noen pasienter har fått avbrudd i behandlingen. Enkelte hevder at den gunstige effekten av enzymterapi varer lenge etter avsluttet behandling (24), men også dette er det uenighet om (25, 26). De fleste synes å være enige om at enzymbehandling ikke bør avbrytes før puberteten av hensyn til skjelettveksten (24 – 26).
Det kan være vanskelig å avgjøre når man skal sette i gang med den meget kostbare og nokså brysomme enzymbehandlingen. Det viktigste behandlingsmålet er å forebygge invalidiserende og irreversible skjelettforandringer. Det bør derfor foretas grundig undersøkelse av skjelettet når diagnosen stilles, med konvensjonell teknikk supplert med mer sensitive metoder som CT og spesielt MR. Pasientene bør følges med nye undersøkelser, og dersom det er tegn til hurtig progrediering, bør røntgenundersøkelser foretas årlig. Fallende verdier for hemoglobin, nøytrofile granulocytter og blodplater kan også utgjøre behandlingsindikasjon. Alvorlig skjelettsykdom kan utvikle seg uten forverring av blodverdiene, og omvendt (20). Ettersom det er så få pasienter i Norge, vil ingen sykehus få stor erfaring. Det kan derfor være klokt å diskutere enkeltpasienter med utenlandske eksperter. Selv har vi hatt stor nytte av kontakt med eksperter i Sverige og Israel.
Behandlingstilbudet til pasienter med type 2- og type 3-sykdom er mer begrenset, selv om utvalgte personer responderer på enzymsubstitusjon. Det foreligger sparsom dokumentasjon på at allogen beinmargstransplantasjon kan gi bedring (27). Dette er et kostbart behandlingsalternativ med en ikke-neglisjerbar morbiditet og mortalitet. Det kan derfor stilles etiske spørsmål om slike transplantasjoner skal tilbys pasienter med type 1-formen, der jo enzymterapi er effektivt. Ingen pasienter med Gauchers sykdom er blitt transplantert i Norge (L. Brinch og A. Glomstein, personlige meddelelser). Nylig har man startet fase 1-studier i den hensikt å evaluere bruk av substratrestriksjon ved Gauchers sykdom type 1. Dette innebærer at man farmakologisk prøver å forhindre akkumuleringen av lysosomale sfingolipider ved å hemme bestemte trinn i deres syntese (28). Endelige data fra disse studiene foreligger imidlertid ikke.
Man har i lengre tid antatt at den optimale terapi må være å erstatte det defekte genet hos pasienter med Gauchers sykdom. Genet for glukocerebrosidase er klonet. In vitro har det vært mulig å overføre dette genet til makrofager og fibroblaster dyrket fra personer med Gauchers sykdom. Med retrovirusvektorer har man også klart å overføre det humane genet til hematopoetiske stamceller hos mus (29). Muligheten for kurativ behandling er altså til stede, men genterapi for denne sykdommen ligger nok et stykke inn i fremtiden.
Det er viktig å diagnostisere og behandle pasientene tidlig for å forebygge senere plager, idet disse menneskene ubehandlet har klart nedsatt livskvalitet som følge av sine organplager (30). I april 2000 gav Rikstrygdeverket for første gang tillatelse til forskrivning av rekombinant glukocerebrosidase til en pasient med Gauchers sykdom for folketrygdens regning.
