Mer enn 95 % av epileptiske anfall stopper spontant i løpet av 1 – 2 minutter. I sjeldne tilfeller ser man at anfallet varer i mer enn 30 minutter. Man taler da om status epilepticus, en alvorlig medisinsk tilstand som klinisk ytrer seg som vedvarende eller gjentatte epileptiske anfall. Alle typer anfall kan utvikle seg i en slik retning, og det er vanlig å skille mellom konvulsiv og ikke-konvulsiv status epilepticus. Status epilepticus av generaliserte tonisk-kloniske anfall, dvs. gjentatte krampeanfall uten oppvåkning mellom anfallene, er mest kjent og fryktet. Status epilepticus av enkle partielle anfall med motoriske symptomer, kalt epilepsia partialis continua, er mindre kjent. Typisk for tilstanden er vedvarende fokale kloniske rykninger i noen muskelgrupper i et lite område av kroppen hos pasienter med bevart bevissthet (1). Rykningene kan vare i timer, dager, uker, måneder og endog år.
Hensikten med denne artikkelen er å rette oppmerksomhet mot denne interessante epilepsiformen og diskutere årsaker, kliniske ytringsformer, elektroencefalografiske og bildediagnostiske funn og differensialdiagnoser. Til slutt drøftes behandlingsmuligheter.
Materiale og metode
I løpet av en toårsperiode har vi ved Spesialsykehuset for epilepsi behandlet 12 pasienter med diagnosen epilepsia partialis continua. Vi har retrospektivt gjennomgått deres journaler og har også funnet relevant litteratur. Demografiske og kliniske pasientdata er vist i e-tabell 1.
|
Tabell 1 Demografiske og kliniske data for 12 pasienter med epilepsia partialis continua
|
|
Pasient 1
|
Pasient 2
|
Pasient 3
|
Pasient 4
|
Pasient 5
|
Pasient 6
|
Pasient 7
|
Pasient 8
|
Pasient 9
|
Pasient 10
|
Pasient 11
|
Pasient 12
|
|
Alder (år)/kjønn
|
46/kvinne
|
60/mann
|
72/kvinne
|
16/kvinne
|
11/mann
|
37/kvinne
|
40/kvinne
|
34/kvinne
|
76/mann
|
3/kvinne
|
67/mann
|
12/kvinne
|
|
Alder ved symptomdebut (år)
|
2
|
54
|
64
|
3
|
10
|
6
|
4
|
20
|
73
|
2
|
28
|
2
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Etiologi,MR-funn
|
Kortikal dysplasi, venstre frontosentroparietalregion
|
Gliom, høyre frontoparietalregion
|
Hjerneinfarkt, høyre sentralregion
|
Schizencefali, venstre frontoparietalregion
|
Rasmussens syndrom. Atrofi av hele høyre hemisfære
|
Artrogryposis multiplex congenita. Schizencefali, venstre parietalregion
|
Ukjent. Cerebral MR: Negativ
|
Gliom, venstre frontoparietalregion
|
Hjerneinfarkt høyre insulaområde
|
Ukjent. Cerebral MR: Negativ
|
Arteriovenøs malformasjon med blødning i venstre frontoparietalregion
|
Encefalitt (Rasmussens syndrom?)Global atrofi, særlig av høyre hemisfære
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Anfallsmønster
|
Vedvarende
|
Intermitterende. Rykninger i 2 – 3 uker. Opptil 3 md. anfallsfrie perioder
|
Intermitterende. Rykninger i 1 – 2 dager. Opptil 2 md. anfallsfrie perioder
|
Vedvarende
|
Intermitterende. Rykninger i flere dager. Opptil 2 – 3 uker anfallsfrie perioder
|
Intermitterende. 3 – 4 anfallsperioder à 30 – 40 min/uke, etterfulgt av Todds parese
|
Intermitterende. 2 – 3 anfallsperioder à 30 – 120 min/uke
|
Intermitterende. 2 – 3 anfallsperioder à 1/2 – 5 timer/md. etterfulgt av Todds parese
|
Intermitterende. Anfallsperioder à 1/2 – 1 døgn ca. hver 3. md.
|
Vedvarende
|
Intermitterende. 1 – 2 anfallsperioder à 5 – 30 min/md.
|
Intermitterende. Anfallsperioder à 2 – 3 døgn hver uke
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Lokalisasjon av rykningene
|
Høyre hånd (ev. hele høyre side)
|
Venstre tunge-, hals- og ansiktshalvdel
|
Venstre hånd (ev. hele venstre side)
|
Høyre hånd (ev. hele høyre side)
|
Venstre fot (ev. hele venstre side)
|
Høyre hånd (ev. hele høyre side)
|
Høyre hånd og ansiktshalvdel (ev. hele høyre side)
|
Høyre hånd og ansiktshalvdel
|
Venstre ansiktshalvdel, (ev. hele venstre side)
|
Venstre hånd og arm
|
Høyre hånd og arm
|
Venstre hånd og arm (ev. hele venstre side)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stimulussensitiv
|
Flimrende lys
|
Bli skremt
|
–
|
–
|
–
|
–
|
–
|
Bli skremt
|
–
|
–
|
–
|
–
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Andre anfallstyper
|
Nei
|
Nei
|
Komplekse partielle anfall, generaliserte tonisk-kloniske anfall
|
Generaliserte tonisk-kloniske anfall
|
Generaliserte tonisk-kloniske anfall
|
Komplekse partielle anfall, generaliserte tonisk-kloniske anfall
|
Komplekse partielle anfall, generaliserte tonisk-kloniske anfall
|
Generaliserte tonisk-kloniske anfall
|
Nei
|
Nei
|
Generaliserte tonisk-kloniske anfall
|
Komplekse partielle anfall, generaliserte tonisk-kloniske anfall
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Effekt av antiepileptika
|
Nei
|
Nei
|
Nei
|
Nei
|
Nei
|
Nei
|
Nei
|
Nei
|
Nei
|
Nei
|
Nei
|
Nei
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Effekt av benzodiazepiner
|
Nei
|
Nei
|
Ja
|
Nei
|
Nei
|
Ja
|
Nei
|
Nei
|
Ja
|
Nei
|
Nei
|
Ja
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Epileptiforme forstyrrelser i EEG under anfall
|
Ja
|
Ja
|
Nei
|
Ja
|
Ja
|
Ja
|
Ja
|
Ja
|
Ja
|
Ja
|
Ingen iktal registrering
|
Ja
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Kirurgi
|
Nei
|
Partiell lesjonektomi
|
Nei
|
Multiple subpiale transeksjoner + frontal reseksjon
|
Hemisfærotomi
|
Nei
|
Nei
|
Partiell lesjonektomi
|
Nei
|
Nei
|
Nei
|
Vagusnervestimulering
|
Resultater
Pasientene, åtte kvinner og fire menn, var i alderen 3 – 76 år. Hos ti av de 12 ble det ved MR av hjernen funnet lesjoner nær sentralfuren (perirolandiske lesjoner) – tre hadde kortikale dysplasier, to hadde gliomer, to hadde hjerneinfarkt, én hadde hatt blødning fra en arteriovenøs malformasjon og to hadde kliniske funn og MR-forandringer mest forenlig med Rasmussens syndrom (en kronisk fokal encefalitt). Epilepsien hadde vart fra ett år til 44 år. Ni pasienter hadde et intermitterende anfallsmønster, tre hadde vedvarende anfall. Hos 11 av pasientene var rykningene lokalisert til ansikt og/eller hånd, og åtte av de 12 hadde andre anfallsformer i tillegg til epilepsia partialis continua.
Ingen ble anfallsfrie med antiepileptika, men fire av de 12 hadde effekt av benzodiazepiner. Det ble funnet iktal epileptiform aktivitet i EEG hos ti av pasientene. Fem har gjennomgått epilepsikirurgi – hos de to med tumor var det utført partiell lesjonektomi, men begge fortsatte å ha intermitterende epilepsia partialis continua etter inngrepet. En av pasientene med kortikal dysplasi ble behandlet med partiell lesjonektomi kombinert med multiple subpiale transeksjoner. Hun fikk færre anfallsepisoder, men ble ikke anfallsfri. Én pasient med Rasmussens syndrom ble helt anfallsfri etter at han omtrent ett år etter sykdomsdebut fikk utført en funksjonell hemisfærotomi. Den andre pasienten med sannsynlig Rasmussens syndrom fikk anfallsreduksjon, men ble ikke anfallsfri etter implantasjon av vagusstimulator.
Fordi pasient 1 anses å være særlig illustrerende og interessant, gjengis hennes sykehistorie og funn mer detaljert.
Pasient 1. Kvinne, født i 1958 etter ukomplisert svangerskap og fødsel. Hun hadde forsinket psykomotorisk utvikling, bl.a. gikk hun ikke før i treårsalderen. Det ble tidlig klart at hun var venstrehendt og at finmotorikken i høyre hånd var svekket. Fra hun var seks måneder til hun var to år hadde hun gjentatte feberkramper. Det er ingen opplysninger om fokale trekk i anfallsutformingen. I toårsalderen begynte hun å få daglige rykninger i høyre kroppshalvdel, uten tap av bevissthet. Disse rykningene har vedvart siden, dvs. i 44 år. Rykningene er mest uttalt i høyre hånd, men kan også forekomme i høyre bein og høyre ansiktshalvdel. Rykningene er ikke til stede under søvn. Vanligvis er det ca. fem rykninger/minutt, men ved stress, søvnmangel eller når hun utsettes for flimrende lys, øker frekvensen til 10 – 20 rykninger/minutt. Hun har aldri hatt sekundærgeneraliserte tonisk-kloniske anfall. Trass i de vedvarende rykningene er hun helt selvhjulpen i daglige gjøremål. Hun er gift og har to friske barn. I 24 år var hun ansatt ved en fabrikk, men for tiden er hun arbeidsledig. Rykningene gjør at hun nødig bærer noe med høyre hånd. Hvis hun gjør det, mister hun ikke det hun bærer som følge av rykningene, men hun kan søle kaffe, suppe o.l. Før hun ble henvist til Spesialsykehuset for epilepsi hadde hun av sin lokale nevrolog fått diagnosen lett cerebral parese med tremor i høyre hånd.
Antiepileptika i form av fenobarbital, fenytoin, karbamazepin og topiramat i doser som gav serumkonsentrasjoner i den øvre delen av referanseområdet, hadde ingen effekt på anfallsfrekvensen. Forsøk på å seponere karbamazepin, som hun for tiden bruker, gav imidlertid forsterkede rykninger. 2 mg klonazepam, gitt rektalt under en EEG-registrering, hadde ingen sikker effekt, verken på hennes kliniske tilstand eller på de elektroencefalografiske forstyrrelsene.
Ved klinisk undersøkelse fant vi moderat hypotrofi og lett sentral parese i høyre arm og hånd.
26-kanalers digital skalp-EEG og videotelemetri viste en 11 – 12 Hz bakgrunnsaktivitet med høyere amplitude av bakgrunnsaktiviteten over venstre hemisfære i forhold til høyre. I venstre frontosentroparietalregion var det hyppig opptredende skarpe bølger («spikes» og «sharp-waves»). Fra tid til annen forekom det paroksysmer av høyspente generaliserte «spike slow-wave»-komplekser med maksimum bifrontosentroparietalt, etterfulgt av elektrodekrement. Disse utbruddene samsvarte med rykninger i høyre hånd (fig 1).
Cerebral MR viste en lesjon subkortikalt i hvit substans frontoparietalt på venstre side. Lesjonen hadde overveiende høyt signal på FLAIR-sekvens (fig 2a) og lavt signal på T1 IR (fig 2b). Lesjonen var traktformet i koronalplanet, med tilspissing ned mot sideventrikkelen. Skillet mellom grå og hvit substans var utvisket. Lesjonen ladet ikke opp kontrast, og størrelsen var uendret sammenholdt med en MR-undersøkelse fra året før. MR-funnene var mest forenlig med en fokal kortikal dysplasi og samsvarer med tidligere publiserte kriterier for denne diagnosen (2). Proton-MRS av lesjonen viste moderat økt kolinnivå og kolin-kreatin-ratio samt redusert N-acetylaspartatnivå og N-acetylaspartat-kreatin-ratio sammenliknet med spektre fra normalt hjernevev i den andre hemisfæren (fig 3). Opptaket er gjort med Chemical Shift Imaging (CSI), PRESS (point-resolved spectroscopy)-sekvens, med ekkotid på 135 ms.
Selv om kvinnen opplever rykningene som sjenerende og frustrerende, særlig i sosiale sammenhenger, ønsker hun ikke å ta imot vårt tilbud om å prøve ut flere antiepileptika (f.eks. valproat eller levetiracetam) eller forsøke operasjon (f.eks. multiple subpiale transeksjoner). «Rykningene er blitt en del av meg.»
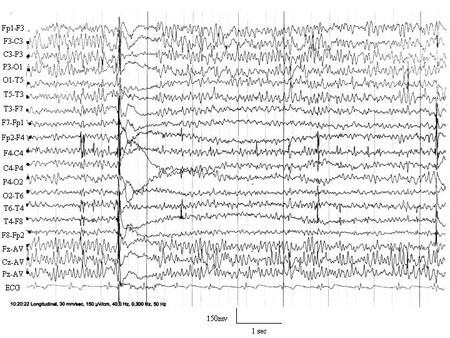
Figur 1 EEG fra pasient 1 (bipolar avledning) viser en amplitudeasymmetri med høyere amplitude over venstre frontosentroparietalregion sammenliknet med høyre side. I tillegg til «spikes» og «sharp waves» i venstre frontosentroparietale region ses korte, generaliserte utbrudd av «spike wave»-komplekser av høy amplitude, fulgt av elektrodekrement. Utbruddene samsvarte med pasientens rykninger i høyre hånd (kurven er vist med redusert sensitivitet; 150 µV/cm)

Figur 2 a) Cerebral MR med koronal FLAIR viser en traktformet hyperintens lesjon i hvit substans frontoparietalt i venstre hemisfære. b) Cerebral MR med koronal T1 IR viser den samme lesjonen med overveiende lavt signal i hvit substans, med typisk avsmalning mot sideventrikkelen

Figur 3 MR-spektroskopi fra lesjonen i venstre frontoparietalregion viser moderat forhøyet kolin- og redusert N-acetylaspartatnivå
Diskusjon
Epilepsia partialis continua har også vært kalt Kojevnikovs syndrom, etter russeren som i 1895 beskrev en epidemisk forekomst av vedvarende fokalmotoriske rykninger hos pasienter som nylig hadde gjennomgått en febersykdom (muligens flåttbåren russisk vår- og sommerencefalitt) (3). Bancaud (4) har foreslått å skille mellom Kojevnikovs syndrom type 1, en ikke-progredierende tilstand som kan ramme mennesker i alle aldre, og Kojevnikovs syndrom type 2, ofte brukt synonymt med Rasmussens syndrom, en progredierende encefalitt som rammer barn. Prevalensen av epilepsia partialis continua er ikke kjent, men tilstanden anses å være sjelden (5).
Epilepsia partialis continua skyldes kortikale og/eller subkortikale lesjoner i de perirolandiske områdene av hjernen. De vedvarende rykningene skyldes først og fremst en affeksjon av primærmotoriske områder, men fordi ca. 40 % av kortikospinale og kortikobulbære fibrer tar utgangspunkt i sensorisk cortex, dvs. bak sentralfuren, kan epilepsia partialis continua også ses ved parietale lesjoner (6). Hvorledes lesjoner i sensomotorisk cortex kan gi opphav til epileptiske forstyrrelser som varer i timer, dager, måneder og år, er ikke fullt ut forstått. Kanskje kan subkortikale lesjoner i disse områdene bevirke en deafferentering av den overliggende cortex, som dermed blir hypereksitabel (1).
Hos de fleste pasienter med epilepsia partialis continua finnes et morfologisk substrat nær sentralfuren. Lesjonene kan være av forskjellig natur (ramme 1) og kan som oftest påvises ved MR-undersøkelse av hjernen, slik som hos ti av våre 12 pasienter. Vanligst er infeksjoner, tumorer og cerebrovaskulære insulter (7). Ved hjelp av de siste års avanserte MR-teknikker er kortikale dysplasier påvist som årsak hos stadig flere pasienter (8), jf. pasient 1, pasient 4 og pasient 6. Hos to av våre pasienter (pasient 7 og pasient 10) ble det ikke funnet noen bakenforliggende årsak, men ingen av disse hadde vært undersøkt med avanserte MR-teknikker. I slike idiopatiske tilfeller mistenkes små kortikale dysplasier, som hos enkelte først avdekkes ved histopatologiske undersøkelser. Hyppigste årsak hos barn og unge er Rasmussens syndrom, jf. pasient 5 og pasient 12. 56 % av personer med dette syndromet utvikler epilepsia partialis continua (9). Etter hvert som den affiserte hemisfæren blir tiltakende atrofisk, blir det økende nevrologiske og kognitive utfall (10). Årsaken til denne progredierende fokale atrofien er ikke kjent, men det har vært postulert både infeksiøse og autoimmune mekanismer (11). Eksempelvis er det hos noen pasienter påvist antistoffer mot en glutamatreseptor (GluR3) (12).
Årsaker til epilepsia partialis continua
Infeksjoner (f.eks. Rasmussens encefalitt, subakutt skleroserende panencefalitt, Creutzfeldt-Jakobs sykdom)
Cerebrovaskulære sykdommer
Tumorer
Kortikale dysplasier
Traumer
Mitokondriell encefalopati
Metabolske forstyrrelser (f.eks. ikke-ketotisk hyperglykemi med hyponatremi, renal og hepatisk encefalopati)
Multippel sklerose
Medikamenter (f.eks. penicillin, metrizamid)
Godartet barneepilepsi med rolandiske «spikes»
Både i de motoriske og de sensoriske barkområdene er det en somatotopisk ordning, og fordi ansikt og hånd dekker betydelige områder, er det ikke overraskende at de fokale rykningene ved epilepsia partialis continua oftest forekommer i ansiktet eller hånden (13). Hos de fleste pasienter holder rykningene seg til de samme muskelområdene over tid, men i sjeldne tilfeller ses en spredning, i en ekstremitet i proksimal retning («Jacksonian march»). Når perioden med rykninger er over hos pasientene med den intermitterende formen, ses ofte forbigående pareser av de muskelgrupper som var gjenstand for rykninger (Todds pareser). De fokale rykningene kan være til stede også under søvn, og hos noen er de stimulussensitive, slik som hos tre av våre pasienter.
Epilepsia partialis continua kan opptre som en forbigående, intermitterende eller vedvarende tilstand. Hos pasienter med den forbigående formen kan tilstanden brenne ut etter en tid. Hos dem med den intermitterende formen kan man se anfallsperioder som varer timer, dager eller uker, med korte eller lange anfallsfrie perioder innimellom. Hos noen ses en eiendommelig regularitet i disse «anfallsbygene», slik som hos pasient 2, som gjennom flere år hadde hatt anfallsperioder som varte 17 – 18 dager, hvorpå han fikk anfallspauser på ca. åtte uker.
At de vedvarende lokaliserte rykningene skyldes epileptiske forstyrrelser i kortikale strukturer, er ikke alltid klinisk innlysende. EEG vil som oftest, men ikke alltid, vise fokale epileptiske forstyrrelser over de affiserte områdene. Er imidlertid lesjonen liten i utbredelse, kan det mangle korrelat i EEG fra elektroder utenpå skallen (standard-EEG), jf. pasient 3. Ved elektrodeplassering innenfor skallen ses imidlertid epileptiform aktivitet, noe som viser at rykningene har kortikal opprinnelse (14). Klinisk kan det av og til være vanskelig å skille tilstanden fra tremor, ekstrapyramidale dyskinesier, ikke-kortikale myoklonier, fleksjonsspasmer og psykiske, ikke-epileptiske anfall (7).
Prognosen ved epilepsia partialis continua beror først og fremst på den tilgrunnliggende årsaken. Ved enkelte tumorformer og alvorlige cerebrovaskulære insulter er prognosen dårlig. Ved stasjonære lesjoner, som f.eks. kortikale dysplasier, ses som oftest ingen nevrologisk eller kognitiv forverring, selv om de fokale rykningene kan vedvare i årevis, slik som hos pasient 1. I to studier hadde tilstanden vart i mer enn ett år hos fire av 21 pasienter i den ene (14) og hos to av 20 i den andre (15).
Epilepsia partialis continua er lite påvirkelig av antiepileptika (1), men kanskje kan slike medikamenter hindre sekundær generalisering til tonisk-kloniske anfall. Benzodiazepiner stopper sjelden de pågående rykningene (14). Hvis rykningene etter uker og måneder ikke opphører, bør man vurdere operasjon. Ved avgrensede lesjoner kan det være aktuelt med en lesjonektomi under kortikografisk veiledning, gjerne supplert med peroperativ kartlegging av motoriske funksjoner ved hjelp av stimuleringsteknikker, men risikoen for å skade elokvent cortex er stor. Et godt alternativ kan være multiple subpiale transeksjoner. Denne metoden går ut på å lage 4 – 5 mm dype kortikale kutt med 5 mm mellomrom i de affiserte barkområdene (16). Ved å skjære over de horisontale intrakortikale fibrene og bevare de vertikale, hindres den epileptiske aktivitet i å spre seg, samtidig som funksjonen i området bevares. Det er nylig rapportert om et godt resultat av multiple subpiale transeksjoner hos en pasient med epilepsia partialis continua forårsaket av en kortikal dysplasi (17).
Ved Rasmussens syndrom bør man vurdere funksjonell hemisfærotomi tidlig i sykdomsforløpet, før de kognitive utfallene er blitt omfattende. Pasient 5 fikk utført en funksjonell hemisfærotomi ca. ett år etter sykdomsdebuten. Han har senere vært anfallsfri.
