Betaoksidasjonen av fettsyrer i mitokondriene består av en serie reaksjoner som bryter ned fettsyrene, og frigjør energi. Den første medfødte defekten i mitokondriell fettsyrenedbrytning ble beskrevet i 1973 (1). Siden er det beskrevet mange defekter (ramme 1), og samlet utgjør de en av de største gruppene av medfødte stoffskiftesykdommer (2). De kan være svært alvorlige, men kan ofte behandles effektivt. Derfor er det viktig å ha et visst kjennskap til disse sykdommene, som kan ramme både barn og voksne.
Ramme 1
Beskrevne metabolske defekter relatert til mitokondriell fettsyrenedbrytning
Karnitinsyklus
Karnitintransport over plasmamembranen
Karnitinpalmityltransferase-I (leverform)
Karnitin-/acylkarnitintranslokase
Karnitinpalmityltransferase-II
Transport av langkjedede fettsyrer over plasmamembranen
Enzymer i betaoksidasjonen
Acyl-CoA-dehydrogenaser for
-
Lange
fettsyrer (VLCAD)
-
Mellomlange
fettsyrer (MCAD)
-
Korte
fettsyrer (SCAD)
3-hydroksyacyl-CoA-dehydrogenaser for
-
Lange
fettsyrer (LCHAD)
-
Korte
fettsyrer (SCHAD)
Tiolase for
Trifunksjonelt protein (TFP)
2,4-Dienyl-CoA-reduktase (hjelpeenzym i nedbrytning av umettede fettsyrer)
Andre
Overføring av elektroner fra acyl-CoA-dehydrogenasene til respirasjonskjeden
Defekter i syntesen av ketonlegemer
Oksidasjon av fettsyrer
Oksidasjon av fettsyrer kan skje både i mitokondriene og peroksisomene. I mitokondriene brytes fettsyrene suksessivt ned til acetyl-CoA. I peroksisomene skjer en tilsvarende oksidasjon av de lengste, ultralange, fettsyrene som blir forkortet, men ikke helt nedbrutt. Fettsyrene kan også omegaoksideres ved innføring av hydroksylgrupper i motsatt ende av syregruppen. Omegahydroksyfettsyrene kan oksideres videre til dikarboksylsyrer, som så kan forkortes, særlig i peroksisomene. Dikarboksylsyrene utskilles raskt i urinen. Påvisning av økt utskilling i urinen av dikarboksylsyrer og andre fettsyrederivater er viktig i diagnostikken.
Lange fettsyrer trenger karnitin for å komme gjennom den indre mitokondriemembranen (3) (fig 1). Fettsyrene blir først omdannet til acyl-CoA-estere av en acyl-CoA-syntetase. Deretter omdannes de til acylkarnitiner av enzymet karnitinpalmityltransferase-I (CPT-I). En egen karnitin-/acylkarnitintranslokase transporterer acylkarnitinene inn i mitokondriene, hvor de tilbakedannes til acyl-CoA-estere av karnitinpalmityltransferase-II (CPT-II). De middelslange og korte fettsyrene kan passere inn i mitokondriene uavhengig av karnitin.
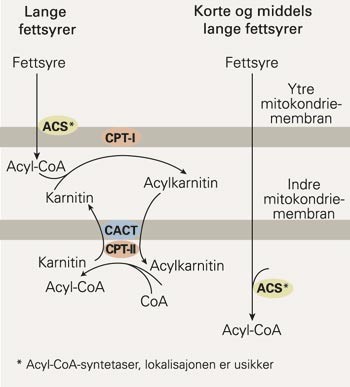
Transport av fettsyrer fra cytosol inn i mitokondriene. Lange fettsyrer transporteres inn ved hjelp av karnitin, mens kortere fettsyrer kan passere direkte inn i mitokondriene. CACT = karnitin-/acylkarnitintranslokase, CPT-I = karnitinpalmityltransferase-I, CPT-II = karnitinpalmityltransferase-II
Selve betaoksidasjonen er en trinnvis avspalting av to og to karbonatomer som acetyl-CoA fra fettsyrekjeden (fig 2). Hver slik forkorting krever fire reaksjoner:
Dehydrogenering, katalysert av acyl-CoA-dehydrogenaser.
Hydrering, katalysert av 2-enyl-CoA-hydrataser.
Dehydrogenering, katalysert av 3-hydroksy-acyl-CoA- dehydrogenaser.
Spalting, katalysert av 3-ketoacyl-CoA-tiolaser.
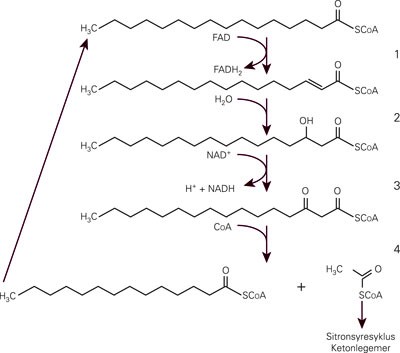
Betaoksidasjon av fettsyrer i mitokondriene. Fettsyrene forkortes med to karbonatomer om gangen. Hver forkorting skjer via fire reaksjoner katalysert av ulike enzymer (1: acyl-CoA-dehydrogenaser; 2: 2-enyl-CoA-hydrataser; 3: 3-hydroksyacyl-CoA-dehydrogenaser; 4: 3-ketoacyl-CoA- tiolaser). For hvert trinn finnes flere enzymer med ulik kjedelengdespesifisitet. Det er tre acyl-CoA- dehydrogenaser, for lange, middels lange og korte fettsyrer, forkortet til henholdsvis VLCAD, MCAD og SCAD. En fjerde dehydrogenase, LCAD, som man opprinnelig trodde var aktiv mot lange fettsyrer, har siden vist seg å ha andre funksjoner. Det er to enyl-CoA-hydrataser og to 3-hydroksyacyl-CoA-dehydrogenaser (LCHAD og SCHAD) for lange og korte fettsyrer. Det er tre tiolaser for henholdsvis lange, middels lange og korte fettsyrer. Acetyl-CoA, som dannes i reaksjonen, kan forbrennes videre i sitronsyresyklus eller omdannes til ketonlegemer i leveren. FAD = flavin adenin dinukleotid, NAD = nikontinamid adenin dinukleotid
Hver av de fire reaksjonene kan katalyseres av flere enzymer med ulik kjedelengdespesifisitet (fig 2). Det er beskrevet fire acyl-CoA-dehydrogenaser, to enoyl-CoA- hydrataser, to 3-hydroksyacyl-CoA-dehydrogenaser og tre tiolaser. De tre siste enzymene i nedbrytningen av lange fettsyrer er lokalisert i samme proteinkompleks, mitokondrielt trifunksjonelt protein.
Acetyl-CoA som dannes i betaoksidasjonen, kan forbrennes videre i sitronsyresyklus eller omdannes til ketonlegemer i leveren. Ketonlegemene kan eksporteres til andre organer og forbrennes der for å skaffe energi.
Fett som energikilde er spesielt viktig ved faste og økt energibehov. Hjerte og muskulatur forbrenner imidlertid fettsyrer også utenom faste. De første timene man faster, er glykogen en viktig energikilde, men etter 12 – 24 timer dekkes > 80 % av energibehovet hos normale barn av fett. Hjernen kan dekke opptil 75 % av sitt energibehov ved forbrenning av ketonlegemer, som også kan forbrennes av hjerte, muskulatur og nyrer.
Fellestrekk ved defekter i mitokondriell fettsyrenedbrytning
Hver defekt har ofte flere kliniske fenotyper. De kan debutere i ulike aldre, fra nyfødtperioden til voksen alder, og er ofte mildest ved debut i voksen alder. Symptomene utløses typisk av infeksjoner, faste eller fysisk aktivitet, dvs. situasjoner hvor fettsyrer er spesielt viktige som energikilde. Symptomene kommer fra organer som i stor grad bruker fett som energikilde, f.eks. hjerte og muskulatur. Dessuten ses ofte leveraffeksjon og encefalopati. Hypoketotisk hypoglykemi er et typisk biokjemisk funn ved de fleste tilstandene. Når cellenes energibehov ikke kan dekkes fra fettsyrer og ketonlegemer, forbrennes glukosen raskere, og glukoseproduksjonen via glukoneogenesen er ikke stor nok til å dekke behovet. Pasientene kan imidlertid være svært dårlige selv med normalt blodsukkernivå, antakelig pga. opphopning av toksiske metabolitter.
Defekter i oksidasjonen av korte fettsyrer kan ha et annet klinisk forløp, f.eks. kan defekt i 3-hydroksy-acyl-CoA- dehydrogenasen for korte fettsyrer gi hyperinsulinisme (4).
Alle sykdommene arves antakelig autosomalt recessivt. De kan være svært alvorlige med høy dødelighet. Prognosen kan imidlertid bedres i betydelig grad ved behandling.
Noen av disse defektene vil bli omtalt nærmere som eksempler. Alle de omtalte defektene er påvist hos norske pasienter, men hyppigheten i Norge er ikke kjent.
Karnitinpalmityltransferase-II-defekt
Karnitinpalmityltransferase-II-defekten var den første defekten i mitokondriell fettsyrenedbrytning som ble beskrevet (1).
Denne tilstanden har to fenotypiske hovedformer. De fleste pasientene har anfall, oftest utløst av fysisk aktivitet, med muskelsmerter og rabdomyolyse. Tilstanden debuterer typisk i alderen 15 – 30 år (5). Pasientene har høy restaktivitet av enzymet. Hos barn er det beskrevet sjeldnere, men alvorligere sykdomsbilder, bl.a. en ofte fatal form med debut i nyfødtperioden eller tidlig barnealder. Pasientene har hypoketotisk hypoglykemi, kardiomyopati og medfødte misdannelser i nyrene og sentralnervesystemet. Misdannelser er ellers ikke typisk for betaoksidasjonsdefekter.
Det finnes mange mutasjoner i genet for karnitinpalmityltransferase-II. En, C439T, finnes i ca. 60 % av allelene hos voksne pasienter med den muskulære formen (6). Mange pasienter er derfor enten homozygote for denne mutasjonen eller sammensatt heterozygote med C439T i ett allel og en annen mutasjon i det andre.
Defekter i nedbrytningen av lange fettsyrer
Defekt i dehydrogenasen for lange fettsyrer (very long-chain acyl-CoA dehydrogenase, VLCAD), ble beskrevet i 1993 (7). Det er påvist en rekke ulike mutasjoner i genet som koder for enzymet, og ingen er spesielt hyppige (8).
De fleste pasientene med defekt i det trifunksjonelle proteinet har en isolert defekt av 3-hydroksy-acyl-CoA- dehydrogenasen, første gang beskrevet i 1989 (9,10). En liten gruppe pasienter har affeksjon av alle enzymaktivitetene i det mitokondrielle trifunksjonelle proteinet. Hos pasientene med isolert 3-hydroksy-acyl-CoA-dehydrogenasedefekt finnes en hyppig punktmutasjon, G1528C (11). Allelfrekvensen av denne mutasjonen hos pasientene er ca. 60 %.
De ulike defektene i nedbrytningen av lange fettsyrer kan være helt like klinisk (12 – 14). Ofte er symptomene utløst av lite matinntak, for eksempel i forbindelse med en infeksjon eller vaksinasjon, som illustrert av følgende kasuistikk.
Pasienten er en nå åtte år gammel pike. Svangerskapet var normalt og fødselen var også normal. Etter DPT- og HIB-vaksinering ved tre måneders alder ble hun gradvis utilpass, slapp, økende blek og hypoton. Hun spiste «ganske greit» inntil siste døgnet, gulpet noe, men hadde ikke oppkast og var afebril. 6 1/2 døgn etter vaksinen ble hun fjern og gav ikke kontakt. Hun ble innlagt i lokalsykehus ved helikoptertransport. Ved innkomst var hun apatisk med åpne øyne og treg pupillreaksjon på lys. Leveren var forstørret til 4 cm under kostalbuen. Hemoglobin 6,6 g/100 ml, natrium 120 mmol/l og blod-glukose 2,6 mmol/l. Maksimal ammoniakk 193 µmol/l (normalt < 50). Spinalvæsken var normal. Man startet behandling med antibiotika, aciklovir og glukose intravenøst. Hun hadde totalt fire anfall med rykninger på høyre side av kroppen.
Hun ble overflyttet til Rikshospitalet med spørsmål om metabolsk sykdom, encefalitt eller Reyes syndrom. Blodprøver her viste moderat leveraffeksjon med maksimalverdi for ASAT 761 U/l, ALAT 267 U/l og bilirubin 66 µmol/l. CK-nivået var forhøyet (537 U/l). Verken vediene for frie fettsyrer eller 3-hydroksybutyrat var forhøyet. Baseoverskudd var – 5,8, laktatnivået lett forhøyet (3,8 mmol/l) og CRP normal. Ultralyd abdomen viste stor lever, mest sannsynlig steatose og noe ascites. Grunnet uklar diagnose ble pasienten leverbiopsert i narkose. Hun hadde dårlig blodsirkulasjon etter prosedyren og ble respiratorbehandlet i to døgn. Ekkokardiografi viste redusert kontraktilitet og røntgen thorax forbigående forstørret hjerte. Leverbiopsien viste uttalt steatose. Urin til metabolsk screening viste svært høye nivåer av patologiske dikarboksylsyrer (fig 3), forenlig med defekt i omsetningen av lange fettsyrer, mest sannsynlig 3-hydroksy-acyl-CoA- dehydrogenasedefekt. Diagnosen ble bekreftet med mutasjonsanalyse som viste at hun var homozygot for den vanlige mutasjonen G1528C. Hun ble satt på fullernæring med en fettfattig morsmelkerstatning med 90 % mellomlange fettsyrer. I tillegg fikk hun essensielle flerumettede fettsyrer i form av omega-3-tran og maisolje, og en lav dose karnitin. Familien fikk nøye informasjon om å unngå faste og å gi glukosepolymerer (for eksempel Semper Energi) ved enhver interkurrent sykdom. Hun begynte raskt med nattlig sondeernæring med pumpe, og ved ett års alder fikk hun anlagt gastrostomi.
Siden har hun vært fulgt opp tett av lege og klinisk ernæringsfysiolog. Dietten har hatt et sterkt begrenset innhold av langkjedet fett. Hun har kun hatt noen få akutte innleggelser i forbindelse med infeksjoner, og aldri alvorlige metabolske kriser. CK har vært en meget følsom markør for hennes metabolske tilstand, med verdier over 10 000 U/l ved interkurrente infeksjoner, uten at hun klinisk har virket særlig påvirket. Snaut fire år gammel fikk hun påvist beskjedne pigmentforandringer i retina, men synet er ikke påvirket. Senere ekkokardiografiske undersøkelser har vært normale. Hun har utviklet seg meget tilfredsstillende og er fysisk sterk uten tegn til nevropati. Hun bør likevel unngå utholdenhetsidretter.
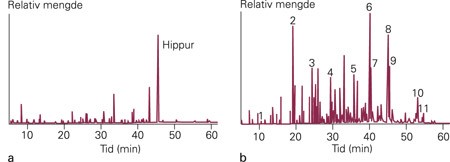
Organiske syrer i urin fra normalperson og pasient med defekt av langkjedet 3-hydroksy-acyl-CoA-dehydrogenase (LCHAD). De organiske syrene er ekstrahert med eter og metylert og deretter separert og identifisert med gasskromatografi-massespektrometri. a) Urin med et tilnærmet normalt mønster av organiske syrer. Hippursyre finnes i størst mengder. b) Urin fra tre måneder gammel pike med akutt metabolsk krise. Mønsteret av organiske syrer er ikke helt spesifikt, men utskillingen av store mengder 3-hydroksydikarboksylsyrer og nærmest ikke ketonlegemer, gir mistanke om LCHAD-defekt. Dette enzymet katalyserer omdanningen fra 3-hydroksy- til 3-ketoacyl-CoA-estere. Når det er defekt, transporteres 3-hydroksy-fettsyrene ut av mitokondriene og omdannes til 3-hydroksydikarboksylsyrer, som så skilles ut i urinen. Bare topper med diagnostisk informasjon er avmerket. 1 3-hydroksybutyrat; 2 Adipinsyre; 3 Korksyre; 4 Dekandisyre; 5 3-hydroksyoktandisyre; 6 3-hydroksydekandisyre; 7 3-hydroksydekendisyre; 8 3-hydroksydodekandisyre; 9 3-hydroksydodekendisyre + hippursyre; 10 3-hydroksytetradekandisyre; 11 3-hydroksytetradekendisyre
Andre fenotyper kan også ses ved disse tilstandene, bl.a. en form karakterisert av myopati og myoglobinuri utløst av anstrengelse eller faste hos voksne, som ved klassisk karnitinpalmityltransferase-II-defekt.
Defekter i det trifunksjonelle proteinet har noen særtrekk, bl.a. et sykdomsbilde med kronisk progredierende polynevropati og myopati hos barn (15). Progredierende retinitis pigmentosa er en vanlig komplikasjon. Akutt åndenød (respiratory distress) er også beskrevet (16). Heterozygote gravide med et affisert foster, er utsatt for akutt fettlever og HELLP-syndrom (17).
Prognosen ved disse sykdommene er dårlig i mange materialer (12, 18, 19). I disse materialene er imidlertid diagnosen ofte først stilt post mortem, og prognosen kan bedres ved tidlig diagnose og riktig behandling (20).
Hovedprinsippet i behandlingen er at pasientene må unngå faste, barn også om natten. Kosten må være karbohydratrik og fettfattig med tilstrekkelig innhold av essensielle fettsyrer. Ukokt maisstivelse (maisennamel) omsettes langsomt og kan brukes for å øke perioden pasientene kan klare seg uten mat. En del av fettet bør inneholde mellomlange fettsyrer, som oksideres av andre enzymer enn de defekte (21). Dette er viktig, ikke minst av hensyn til hjertet, som bruker fett til forbrenning. Man håper at et slikt tilskudd kan forhindre kardiomyopati. Om L-karnitin skal gis er omdiskutert, og det anbefales seponering av eventuelt tilskudd ved akutt metabolsk krise. Ved infeksjoner er det viktig å hindre katabolisme og fettsyremobilisering. Foreldrene må derfor ha en egen skriftlig behandlingsplan (kalt SOS-regime) for peroral tilførsel av glukosepolymerer hjemme, og fri tilgang til sykehus ved behov. Det viktigste ved akutt sykehusbehandling er høy energitilførsel (3 360 – 4 200 J (80 – 100 kcal)/kg/døgn). Peroralt kan man gi 10 % av energibehovet som triglyserider med mellomlange fettsyrer (MCT-fett), 75 – 80 % som karbohydrater og 10 – 15 % som proteiner/aminosyrer. Ofte tåles imidlertid rene karbohydratløsninger best. Intravenøst gis 12 – 20 % glukose, ev. med insulin i tillegg, samt aminosyrer (22).
Defekt i nedbrytningen av mellomlange fettsyrer
Defekt i dehydrogenasen for mellomlange fettsyrer (MCAD-defekt) ble første gang beskrevet i 1982 (23), og er den vanligste betaoksidasjonsdefekten i mange land. Sykdommen er særlig hyppig i den kaukasiske befolkningen, og hyppigheten kan være så høy som 1/10 000 nyfødte. Siden 1998 har vi funnet denne defekten hos seks barn, og den er den hyppigste fettsyrenedbrytningsdefekten i vårt materiale.
En vanlig punktmutasjon, A985G (24), utgjør opptil 80 – 90 % av sykdomsallelene, så mange av pasientene, bl.a. fire av våre, er homozygote for denne mutasjonen. Til tross for den ene hyppige mutasjonen varierer den kliniske fenotypen.
En typisk presentasjon av sykdommen ses hos små barn som blir syke etter en periode med faste, ofte pga. infeksjon. De kaster opp, blir letargiske og eventuelt komatøse. De har ofte, men ikke alltid, hypoglykemi og lave til moderate nivåer av ketonlegemer i urinen. Verdien av ALAT er forhøyet. Kronisk muskelaffeksjon og kardiomyopati er ikke vanlig, men akutt kan CK-konsentrasjonen øke kraftig. Sykdommen kan debutere som plutselig uventet spedbarnsdød, men er ikke en hyppig årsak til krybbedød. Alle som har defekten biokjemisk, blir ikke klinisk syke, men symptomene kan vise seg først i voksen alder (25).
Denne defekten er likevel alvorlig. Dette illustreres av et pasientmateriale som omfattet 120 barn fra 96 familier (26). 23 (19 %) av pasientene døde, men ingen etter at diagnosen var stilt. Mange av pasientene som overlevde, hadde dessverre sekveler etter metabolske kriser med symptomer som muskelsvakhet, epilepsi, dårlig trivsel, cerebral parese, språkproblemer og hyperaktivitet.
Ved riktig behandling kan metabolske kriser unngås, eller alvorlighetsgraden av anfallene reduseres. Behandlingen er mindre krevende og rigorøs enn ved defekter i oksidasjonen av langkjedede fettsyrer, fordi pasientene kan ha et vanlig, sunt kosthold med regelmessige måltider i det daglige. Ofte gis ukokt maisstivelse om kvelden, i hvert fall til barn. Behovet for tilskudd av L-karnitin er omdiskutert, men slikt tilskudd gis ofte. Heller ikke disse pasientene tåler langvarig faste. Hvis barnet spiser dårlig, gis karbohydrater (glukosepolymerer, SOS-regime), og sykehusinnleggelse for sondeernæring eller intravenøs ernæring kan bli nødvendig. Behandlingen består av høy energitilførsel (3 360 – 4 200 J (80 – 100 kcal)/kg/dag), med minst 70 % som karbohydrater. Pasientene må ikke få mellomlange fettsyrer! Dessuten gis karnitin, 100 – 200 mg/kg/døgn fordelt på 3 – 4 doser per os eller intravenøst (22).
Diagnostikk av defekter i mitokondriell betaoksidasjon
Diagnostikken av disse tilstandene bygger på klinisk mistanke. Den er enklere ved metabolske kriser enn utenom (2), og spesielt ved milde defekter kan det være vanskelig å stille diagnosen. Det er derfor viktig at urin- og blodprøver (serum og EDTA-plasma) tas tidlig i akuttforløpet, fryses ned og sendes til Avdeling for medisinsk biokjemi, Rikshospitalet. Siden pasientene ikke tåler å faste, må man være svært forsiktig med provosert faste.
Vanlige blodprøver og urinstiks tatt i akuttfasen kan gi mye informasjon. Typiske funn som ses ofte, men ikke alltid er: hypoketotisk hypoglykemi, patologiske leverprøver, økt konsentrasjon av CK, CK-MB og myoglobin, lett metabolsk acidose og moderat forhøyede nivåer av laktat, ammoniakk og urinsyre.
Konsentrasjonen av frie fettsyrer er høy. Ved alle defektene ses lav totalkarnitinkonsentrasjon i serum med redusert fri fraksjon, bortsett fra ved karnitinpalmityltransferase-I-defekten hvor konsentrasjonen av totalkarnitin er økt. Under en akutt metabolsk krise kan imidlertid karnitinverdiene være høye. En medfødt, arvelig defekt i opptaket av karnitin over cellemembranene gir svært lave karnitinverdier både i plasma og intracellulært, og ble tidligere kalt primær karnitinmangel.
Histologisk undersøkelse av biopsier vil ofte vise fettavleiring i lever og muskulatur. Hjertet bør undersøkes med ekkokardiografi.
Ved Seksjon for biokjemisk genetikk undersøker vi organiske syrer i urin og acylkarnitiner i plasma. Mønsteret av organiske syrer i urinen (fig 3) viser økt utskilling av dikarboksylsyrer og hydroksydikarboksylsyrer kombinert med liten utskilling av ketonlegemer, i hvert fall under metabolske kriser. Ved MCAD-defekten ses i tillegg spesielle glysinkonjugater. Acylkarnitinene i plasma er oftere patologiske også utenom akutte episoder med forverring. Vi kan også undersøke fettsyreoksidasjonen in vitro i lymfocytter etter avtale.
Måling av enzymaktivitet kan bekrefte en mistenkt diagnose. I Norge måles kun karnitinpalmityltransferase. For andre diagnoser sendes fibroblaster til utenlandske laboratorier. Mutasjonsanalyser kan også bekrefte en mistenkt diagnose.
I mange land, men ennå ikke i Norge, analyseres acylkarnitiner som en del av nyfødtscreeningen. Derved kan mange betaoksidasjonsdefekter oppdages og behandles før de har gitt symptomer (17).
Konklusjon
Defekter i mitokondriell fettsyreoksidasjon forekommer sjelden, men er viktige å ha kjennskap til, fordi behandling av disse alvorlige sykdommene kan bedre prognosen vesentlig. Tidlig diagnose basert på klinisk mistanke er derfor viktig.
