Det var troponinstigning hos en 58 år gammel kvinne innlagt med hjerneslag. Hun utviklet senere i sykdomsforløpet utbredt tromboembolisme og multiorgansvikt. Den underliggende årsaken representerer en diagnostisk og terapeutisk utfordring.
Se kunnskapsprøve på www.tidsskriftet.no/quiz
En 58 år gammel kvinne med kjent hypertensjon og migrene uten aura ble innlagt ved et sentralsykehus med spørsmål om hjerneslag. Hun brukte ingen faste medisiner. Innleggelsesdagen, dag 1, hadde hun tre dagers sykehistorie med forbigående kraftsvikt i høyre bein, høyresidig hodepine, dobbeltsyn og smerter i venstre arm. Ved klinisk undersøkelse var alt normalt, bortsett fra at blodtrykket var 190/90 mm Hg. EKG viste inverterte T-bølger i V1 og V2 og blodprøvene troponin I 3,26, CK-MB 13 og D-dimer 17,8. Akutt hjerteinfarkt, hjerneslag, lungeembolisme og aortadisseksjon var mulige diagnoser. CT caput og thorax viste intet galt. CT abdomen viste en mulig fortykket vegg i antrum ventriculi samt flere grensestore lymfeknuter. Funnet ble tolket som en normalvariant uten sikker klinisk betydning.
En rekke tilstander kan gi samtidig troponinstigning og hjerneslag. Denne kombinasjonen kan ses ved akutt koronarsyndrom, hvor om lag 1 % utvikler hjerneslag (1). Infeksiøs endokarditt kan også gi troponinstigning, og ett eller flere hjerneinfarkter kompliserer tilstanden i ca. 10 % av tilfellene (2). For pasienter med hjerneslag er det påvist troponingstigning hos 9,6 – 27 % (3, 4). En viktig årsak til myokardskaden hos slagpasienter er trolig sentral hyperaktivering av det sympatiske nervesystem med sekundær katekolamintoksisitet (5). Ellers er hjertesvikt og nyresvikt vanlige tilstander som kan gi troponinstigning. Et hjerneslag hos slike pasienter vil gi den aktuelle kombinasjonen. For øvrig kan aortadisseksjon, vaskulitter, koagulopatier og sepsis affisere både hjerte og hjerne. Hos vår pasient ønsket man i første omgang å avklare om hun hadde koronarsykdom.
Dag 3 ble pasienten overflyttet til et annet sykehus for koronarangiografi. Den var normal. Dag 4 ble hun tilbakeført til sentralsykehuset for videre utredning. Da var hun symptomfri. Hun valgte, på tross av informasjon og råd, å reise hjem for videre utredning hos fastlegen. Hun ble utskrevet med metoprolol 100 mg x 1 og lisinopril 5 mg x 1.
Dag 8 ble hun på nytt innlagt ved sentralsykehuset. Hun var da forvirret og motorisk urolig. Det var multiple ekkymoser på truncus og ekstremitetene. I løpet av neste døgn utviklet hun venstresidig hemiparese, visuell neglekt, hemianopsi og global afasi. Cerebral CT og MR viste infarktutvikling i fremre, midtre og bakre deler av høyre hemisfære, et mindre infarkt i venstre hemisfære samt bilaterale infarkter i cerebellum. Blodprøver viste troponin I > 10, CK-MB 22, CRP 73, hvite blodceller 19, trombocytter 200, fibrinogen 2,6, INR 1,0 og cephotest 27.
Transøsofageal ekkokardiografi viste ingen embolikilder. MR-angiografi av cerebrale og precerebrale kar viste normale forhold. Biopsi av ekkymoser viste ikke vaskulitt. Negative blodkulturer gjorde sepsis mindre sannsynlig. ANA, ANCA, revmatoid faktor, antikardiolipinantistoffer og lupusantikoagulant var negative. Spinalvæsken var normal. Beinmargsutstryk viste reaktiv marg med økt antall megakaryocytter uten tegn til leukemi. Perifert blodutstryk viste ikke tegn til mikroangiopatisk hemolytisk anemi.
Pasienten ble behandlet med cefotaksim 2 g x 3, metronidazol 500 mg x 3, enoksaparin 40 mg x 2 og forsøksvis faktor VIII-konsentrat og metylprednisolon 1 g x 1. Dag 16 ble hun overflyttet til Rikshospitalet. Hun var da paralytisk i venstre kroppshalvdel, desorientert, men våken. Det ble da foretatt en ny MR-undersøkelse (fig 1). Fra dag 20 til dag 25 falt trombocyttallet fra 200 til 23. Innenfor samme tidsrom utviklet hun makroskopisk hematuri og melena. INR og cephotest lå i normalområdene, fibrinogennivået hadde sunket noe, men lå fortsatt i referanseområdet. D-dimer var 19. Man mistenkte heparinindusert trombocytopeni type 2 (HIT2), og enoksaparin ble seponert dag 22. Acetylsalisylsyre 160 mg x 1 ble valgt som eneste tromboseprofylakse.
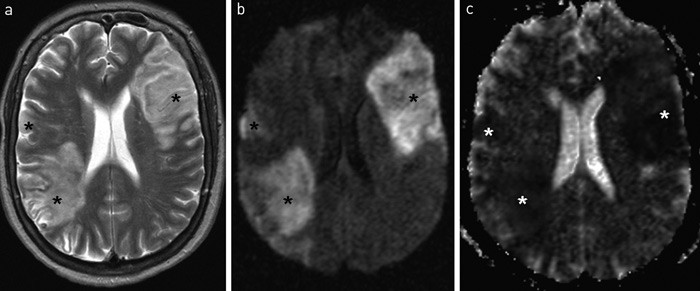
Figur 1 a) T2-vektet cerebral MR viser flere infarkter. b) Diffusjonsvekting viser ulike diffusjonsfunn, og c) ADC-kartet (apparent diffusion coefficient) med forskjellige signalmønstre bekrefter at det er infarkter av ulik alder
Hematuri og melena kunne skyldes blødningstendens på grunn av trombocytopeni, tromboembolisk sykdom som affiserte nyrer og tarm eller intravaskulær trombosering. Heparin danner komplekser med platefaktor 4 (PF4). Heparinindusert trombocytopeni type 2 oppstår når immunforsvaret danner antistoffer mot disse kompleksene. Dette kan gi blødningstendens og livstruende trombotiske komplikasjoner. Selv om dette forekommer svært sjelden ved bruk av lavmolekylære hepariner (6), er det en viktig differensialdiagnose hos heparinbehandlede pasienter med trombocytopeni eller andre koagulasjonsforstyrrelser. Ved heparinindusert trombocytopeni type 2 bør man seponere heparin og behandle med en direkte trombinhemmer (6).
I løpet av dag 29 utviklet pasienten respirasjonssvikt, og bevissthetsnivået sank gradvis. Hun ble intubert og lagt i respirator. Nivået av CRP og hvite blodceller var stigende. Røntgen thorax viste venstresidig pleuravæske og fortetninger i høyre overlapp. CT thorax viste multiple bilaterale lungeembolier. Pasienten fikk cefuroksim 750 mg x 3 grunnet mistanke om sepsis og standard heparin intravenøst. Blodprøver dag 30 viste Hb 6,8 g/100 ml, fibrinogen 0,9, INR 1,5, D-dimer > 20 og trombocytter 53. Blodutstryk var normalt. Aspirat og biopsi fra beinmarg viste som før reaktiv marg med økt antall megakaryocytter.
Ved heparinindusert trombocytopeni type 2 ville pasienten fortsatt hatt antistoffer mot heparin-PF4-komplekser i serum få dager etter seponert heparinbehandling. Ved ny eksponering for heparin ville man forventet et raskt og betydelig fall i trombocyttallet. Da dette tvert imot økte noe, er heparinindusert trombocytopeni type 2 mindre sannsynlig. En kronisk forbrukskoagulopati kan forklare det tidligere observerte fallet i trombocyttall. Denne hypotesen styrkes av høy D-dimer, reaktiv beinmarg, synkende fibrinogenkonsentrasjon og stigende INR-nivå. Det er nærliggende å tro at pasienten tidlig i sykdomsforløpet hadde en kompensert disseminert intravaskulær koagulasjon (DIC). Senere i forløpet overskred forbrukskoagulopatien leverens og beinmargens kompenserende evner, og trombocyttallet falt.
Kreftmarkøren CA 125 var 60 (0 – 35), og CT abdomen fra sentralsykehuset ble gransket på nytt. En mulig fortykket antrum ventriculi og grensestore lymfeknuter fikk ny oppmerksomhet.
CA 125 er et mucinøst glykoprotein som brukes til å følge forløpet ved ovariecancer, men konsentrasjonen kan også øke ved en rekke andre tilstander, inkludert ventrikkelcancer. I en artikkel beskrives fire pasienter med metastatisk mucinøs cancer, hjerneinfarkt og annen tromboembolisk sykdom med forhøyede verdier av CA 125 (7).
CT og MR abdomen viste forverring av de nevnte funnene og infarkter i leveren og begge nyrene (fig 2). Pasienten ble gastroskopert. Biopsi fra ventrikkelen viste et lite differensiert mucinøst adenokarsinom av diffus type. Man konkluderte med at sykdomsbildet skyldtes kreftindusert hyperkoagulabilitet. Kvinnen døde dag 36.
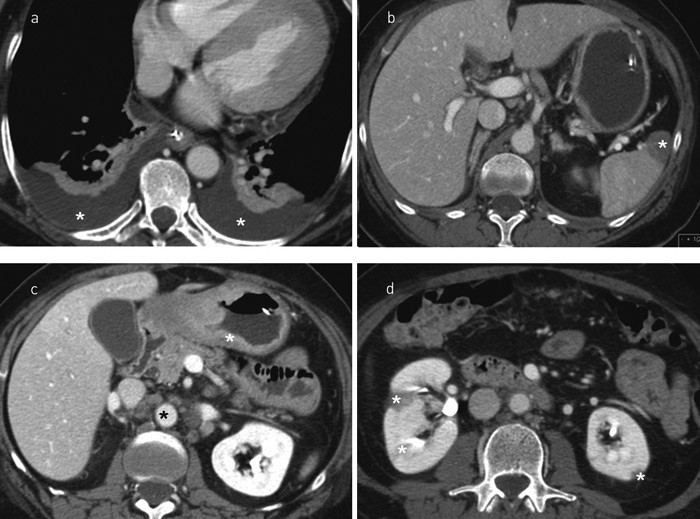
Figur 2 CT thorax og abdomen tatt få dager før pasienten døde. a) Betydelig pleuravæske (hvite stjerner). b) Infarkt i milt (hvit stjerne). c) Betydelig fortykket vegg i ventrikkel (hvit stjerne) og patologisk forstørrede lymfeknuter rundt aorta (svart stjerne). Dette bildet førte til biopsi og endelig diagnose. d) Infarkter i begge nyrer (hvite stjerner)
Diskusjon
Forbindelsen mellom kreft og trombose ble første gang beskrevet av Armand Trousseau i 1865 (8). Han beskrev venøs trombose og residiverende migrerende tromboflebitter hos pasienter med okkult cancer. Nyere litteratur definerer imidlertid Trousseaus syndrom på ulike måter. Vi vil her bruke «Trousseaus syndrom» om symptomgivende venøs og/eller arteriell hyperkoagulabilitet ved erkjent eller okkult kreftsykdom.
Ved Trousseaus syndrom er venøs tromboembolisme (VTE) hyppigere enn arteriell. Venøs trombose ses hos om lag 50 % av kreftpasienter i obduksjonsmaterialer (9). 5 – 15 % av alle kreftpasienter utvikler klinisk venøs tromboembolisme (10, 11). Trousseaus syndrom ses hyppigst ved leukemier og kreft i lunge, pancreas, ventrikkel og colon og ved metastatisk sykdom (11, 12). Sannsynligvis kan alle kreftformer gi hyperkoagulabilitet (9).
Hos pasienter med idiopatisk venøs tromboembolisme er risikoen for å få en kreftdiagnose innen 6 – 12 måneder økt 3 – 4 ganger, og om lag 10 % utvikler kreft innen 5 – 10 år (11, 13). Foreløpig anbefales imidlertid ikke omfattende kreftutredning ved idiopatisk venøs tromboembolisme.
Sannsynligvis er det flere komplekse og samvirkende faktorer som står bak den systemiske hyperkoagulabiliteten som kan opptre hos kreftpasienter. Kreftceller uttrykker protrombotiske faktorer som vevstromboplastin (tissue factor, TF) og cancerprokoagulant. Disse aktiverer koagulasjonskaskaden (10). Enkelte adenokarsinomer frigjør mucin, som kan gi trombedanning (14). Vertens respons på tumor er protrombotisk og inkluderer akuttfasereaksjon, inflammasjon og nekrose. Mononukleære celler interagerer med maligne celler og utskiller IL-1, IL-6 og tumornekrosefaktor. Dette gir endotelskade, trombocytt-, faktor XII- og faktor X-aktivering og trombindanning (10, 15). Indirekte faktorer som immobilisering, kirurgi, intravaskulær kateterbehandling, hormonterapi, kjemoterapi og infeksjoner gir også økt risiko for tromboembolisme.
Kreftindusert hyperkoagulabilitet kan gi et svært variert sykdomsbilde – alt fra en subklinisk protrombotisk tilstand til disseminert intravaskulær koagulasjon, venøs tromboembolisme og ikke-bakteriell trombotisk endokarditt (NBTE). Ved sistnevnte finnes sterile tromber på hjerteklaffene. Trombene kan embolisere til ethvert organ (16). Ikke-bakteriell trombotisk endokarditt ses hos 3 – 4 % av kreftpasienter i større obduksjonsmaterialer (17, 18), og 40 – 50 % av disse har embolisk hjerneinfarkt. Om lag 8 % av dem med ikke- bakteriell trombotisk endokarditt utvikler embolisk hjerteinfarkt (19). Ofte ses embolier til lunger, nyrer og milt.
Pasienter med Trousseaus syndrom har oftere avansert kreftsykdom og høyere dødelighet enn andre kreftpasienter. I en retrospektiv studie fant man en ettårsoverlevelse på 12 % (20). I en prospektiv studie over ett år hadde 20,7 % fått residiv av venøs tromboembolisme, mot 6,8 % i kontrollgruppen (21). Risikoen for slikt residiv er spesielt høy dersom det er avansert kreftsykdom.
Kreftpasienter med venøs tromboembolisme bør behandles med lavmolekylært heparin i 3 – 6 måneder (22). CLOT-studien viste at seks måneder med subkutan injeksjon av et lavmolekylært heparin (dalteparin) var mer effektivt enn vitamin K-antagonist som sekundærprofylakse hos disse (23). Risikoen for alvorlig blødning var den samme som ved peroral antikoagulasjonsbehandling. Det finnes per i dag ingen studier der man har sammenliknet lavmolekylært heparin med peroral antikoagulasjonsbehandling utover seks måneder. Man bør i alle fall behandle med lavmolekylært heparin i mer enn seks måneder ved avansert kreftsykdom, ved venøs tromboembolisme under behandling med vitamin K-antagonist og der tilstanden gjør bruk av perorale antikoagulantia vanskelig (24). Man bør vurdere livslang antikoagulasjonsbehandling eller behandle inntil kreftsykdommen er helbredet (22). Flere studier har vist at behandling med lavmolekylært heparin øker overlevelsen hos kreftpasienter, hovedsakelig gjelder det dem uten metastaser (24).
Kreftpasienter som immobiliseres eller behandles kirurgisk bør få lavmolekylært heparin profylaktisk (25). Pasienter uten slike risikofaktorer bør ikke behandles med heparin, heller ikke pasienter med sentralt venekateter. Venekateterindusert trombose er sjeldnere enn tidligere antatt, og antikoagulasjonsbehandling reduserer forekomsten i liten eller ingen grad (25).
Det finnes i dag ingen etablerte behandlingsstrategier hos kreftpasienter med disseminert intravaskulær koagulasjon og ikke-bakteriell trombotisk endokarditt. De nevnte studier om venøs tromboembolisme og kreft gjør at lavmolekylært heparin kan være et alternativ. Den eneste effektive behandlingen ved disseminert intravaskulær koagulasjon er imidlertid å fjerne årsaken.
Pasienten vår utviklet Trousseaus syndrom med bilateral lungeemboli og infarkter i hjerte, hjerne, lever og nyrer. Vevstromboplastin er en sentral mediator av systemisk hyperkoagulabilitet hos kreftpasienter. Ulike typer maligne svulster kan uttrykke denne faktoren, også adenokarsinomer i ventrikkelen (26). vevstromboplastinmediert hyperkoagulabilitet kan være en felles patofysiologisk mekanisme bak pasientens symptomer og funn. En annen mulig forklaring er emboliserende mucin fra primærtumor eller fra metastaser.
Konklusjon
Trousseaus syndrom forekommer hyppig og medfører betydelig mortalitet og morbiditet hos kreftpasienter. Ved økt risiko for tromboembolisme bør kreftpasienter få tromboseprofylakse i form av lavmolekylært heparin. Pasienter med kreft og venøs tromboembolisme bør behandles med lavmolekylært heparin i minst 3 – 6 måneder.
