En mann i begynnelsen av 80-årene ble lagt inn på lokalsykehuset med spontane og anemiserende hud- og muskelblødninger. Pasienten brukte både warfarin og acetylsalisylsyre, men blødningene viste seg å ha en langt sjeldnere årsak enn medikamentbivirkning.
En mann i begynnelsen av 80-årene ble lagt inn på sitt lokalsykehus med hematom i høyre over- og underarm uten kjent traume. Han var behandlet med warfarin, og INR var målt til over 8,0 hos innleggende lege. Pasienten var bypassoperert og hadde fått innsatt biologisk aortaklaff noen få år tidligere. I etterkant av dette ble det konstatert persisterende atrieflimmer. Grunnet fravær av andre sykdommer, høyt funksjonsnivå og ingen tidligere blødninger ble han behandlet både med warfarin og acetylsalisylsyre. Frem til den aktuelle tilstanden hadde han hatt stabil INR og ingen blødninger.
Hos pasienter med koronar hjertesykdom og atrieflimmer vil man som regel anse behandling med kun acetylsalisylsyre som utilstrekkelig for å redusere risiko for systemisk arteriell embolisering. Det er hos utvalgte pasienter med antatt liten risiko for blødning og god etterlevelse akseptert å behandle både med warfarin og acetylsalisylsyre. Ved kombinasjonsbehandling øker riskoen for blødning 2 – 3 ganger sammenliknet med monoterapi med warfarin (1), og man har antakelig kun gevinst av kombinasjonsbehandling hos pasienter med stabil INR mellom 2,0 og 3,0 (2).
Pasienten fortalte at han hadde hatt økende smerter i proksimal muskulatur og økende muskelstivhet i skuldre og hofter de siste tre måneder forut for innleggelse. Dette hadde ført til redusert funksjonsnivå og problemer med daglige gjøremål. Fire dager før innleggelsen oppsto det spontant et hematom som omfattet distale tredel av overarm og underarm ned til håndleddet. På grunn av smertene fikk han opiater. Pasienten hadde redusert fleksjon og ekstensjon av samtlige fingre og lett nedsatt sensibilitet i underarm og fingre. Sirkulasjon ble vurdert som normal. Blodprøver ved innkomst viste Hb 9,0 g/dl (13,4 – 17,0), trombocytter 409 · 10⁹/l (145 – 348), kreatinin 63 μmol/l (60 – 105), CRP 49 mg/l (< 5), aktivert partiell tromboplastintid (aPTT) 68 s (28 – 42), D-dimer 0,8 mg/l (< 0,5), fibrinogen 5,4 g/l (2,0 – 4,0) og INR 3,2 (0,8 – 1,1). Ved innkomst ble warfarin og acetylsalisylsyre stoppet og pasienten fikk 10 mg K-vitamin. Etter ett døgn var INR falt til 2,2. På dag 3 var INR 1,2, aPTT 68 s, trombocytter 267 · 10⁹, Hb 8,6 g/dl. Det var likevel tiltakende blødning i høyre underarm, med økende bevegelsesavhengige smerter. Ortoped så til han flere ganger for å vurdere eventuell fasciotomi og evakuering av hematom. Man valgte å avstå fra kirurgisk intervensjon. Etter fem dager var INR < 1,5 og underarmomkretsen minket, pasienten hadde mindre smerter og forbedret sensibilitet og motorikk. Han fikk ingen antikoagulasjonsbehandling, men hadde fortsatt spontane hudblødninger. Det ble observert vedvarende blødning og reblødninger fra innstikksteder etter blodprøvetaking. Nivåene av INR og trombocytter var nå normale, men aktivert partiell tromboplastinttid var vedvarende forhøyet > 55 s..
Etter at warfarineffekten var opphevet, hadde pasienten fortsatt uttalt blødningstendens. Det eneste labratoriefunnet var forlenget aktivert partiell tromboplastinttid (aPTT). Aktivert partiell tromboplastinttid er en koagulasjonstest som brukes til å vurdere den interne kaskade (faktor VIII, IX, XI og XIII) og den terminale kaskade (faktor II, V og I) (fig 1). Spontan forlenget aktivert partiell tromboplastinttid kan skyldes medfødt faktormangel, som mangel på faktor VIII, faktor IX eller faktor XII. Medfødt faktor XII-mangel fører til forlenget aktivert partiell tromboplastinttid uten å gi blødningstendens. Medfødt faktor VIII- og faktor IX-mangel gir forlenget aktivert partiell tromboplastinttid og blødningstendens. Siden pasienten tidligere var bypassoperert uten noen postoperativ stor blødning, var medfødt faktormangel svært lite sannsynlig. Den vanligste ervervede tilstanden som gir forlenget aktivert partiell tromboplastinttid er lupusantikoagulans. Hos eldre pasienter er den vanligste årsaken til forlenget aktivert partiell tromboplastinttid lupusantikoagulans eller heparineffekt (3). Lupusantikoagulans er antistoffer som er rettet mot fosfolipidene i aPTT-reagensen og dermed blokkerer aktivering av den interne kaskaden (4, 5). Selv om lupusantikoagulans gir forlenget aktivert partiell tromboplastinttid, er det ikke assosiert med blødning, men med trombose. Spontan forlenget aktivert partiell tromboplastinttid og nyoppstått blødningstendens talte for at pasienten hadde en ervervet faktormangel med autoantistoffer mot enten faktor VIII, faktor IX eller faktor XI.
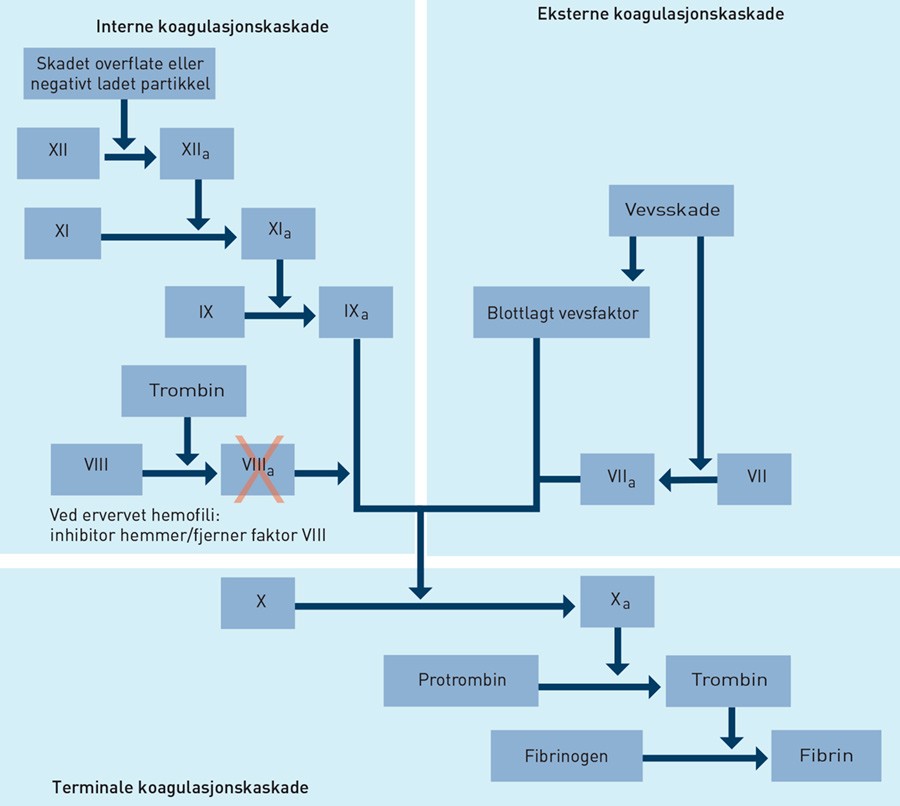
Figur 1 Forenklet fremstilling av koagulasjonskaskaden. Faktor VIII sammen med fosfolipider, faktor IXa og kalsium aktiverer faktor X i den terminale koagulasjonskaskaden. Faktor FVIIIa er en helt essensiell forsterkingsmekanisme for danning av store mengder trombin
Repeterte målinger hos den aktuelle pasienten bekreftet forlenget aktivert partiell tromboplastinttid i fravær av heparin (f.eks. venefloner med heparinlås). Faktoranalyse gjort ved Rikshospitalet viste faktor VIII-nivå på 4 % (50 – 150 %) og tilstedeværelse av en inhibitor mot faktor VIII med inhibitornivå 20 BE (Bethesda-enheter), noe som bekreftet diagnosen ervervet hemofili.
Ervervet hemofili er en tilstand med autoantistoffer mot koagulasjonsfaktorer. Et slikt autoantistoff omtales også som en inhibitor. I om lag 50 % av tilfellene kan man påvise en assosiert lidelse, og da som regel enten en annen autoimmun sykdom eller malignitet (6). Revmatiske sykdommer er rapportert hos 10 – 20 %. De aller fleste med ervervet hemofili og revmatisk lidelse har enten revmatoid artritt eller systemisk lupus erythematosus. Polymyalgia rheumatica er rapportert hos ca. 2 % hos pasienter med ervervet hemofili (6). Blant dermatologiske lidelser ses ervervet hemofili utelukkende ved pemphigus vulgaris og psoriasis. Underliggende malignitet blir funnet hos 2 – 10 %. Det ser ut som ervervet hemofili opptrer hyppigere ved solide tumorer enn ved lymfoproliferative/myeloproliferative tilstander. Det er usikkerhet om ervervet hemofili kan utløses av medikamenter (7).
Ved klinisk undersøkelse fant man uttalt proksimal muskelstivhet, og pasienten hadde problemer med å løfte hendene over hodet og med å sette seg på huk. Arteria temporalis var klinisk normal og biopsi av denne ble ikke utført grunnet blødningsrisiko. Revmatolog konkluderte med diagnosen polymyalgia rheumatica/arteritis temporalis, og at det ikke fantes holdepunkter for annen revmatologisk tilstand. CT av hals, thorax og abdomen ga ikke holdepunkter for underliggende malign tilstand.
Behandling av pasienter med ervervet hemofili består av å stanse blødning ved hjelp av koagulasjonsfaktorer og å eradikere autoantistoffer ved hjelp av immunsuppressiv behandling. Ved medfødt hemofili får pasienten substitusjonsbehandling med faktor VIII. Ved ervervet hemofili vil autoantistoffer også inaktivere tilført faktor VIII. Ved mindre alvorlige blødninger og ved inhibitornivå under 5 BE kan man forsøksvis gi substitusjonsbehandling med høye doser faktor VIII-konsentrat (100 – 200 IE/kg) for å overkomme inhibitoren (5). Best dokumentert effekt ved blødning har tilførsel av aktivert faktor VIIa (NovoSeven) eller FEIBA (Faktor EIght Bypassing Agent), som er et aktivert protrombinkompleks-konsentrat (aPCC) (5, 8). Pasienter med ervervet hemofili og blødninger bør håndteres ved sykehus med tilstrekkelig tilgang på faktorkonsentrat og hematolog med spesialkompetanse innen koagulasjonsforstyrrelser. Eradikering av autoantistoffer oppretter normale faktornivåer og minimaliserer dermed risiko for alvorlig blødning.
På dette tidspunktet hadde pasienten ingen blødning og dermed ikke noe behov for faktorkonsentrat. Man startet eradikasjonsbehandling umiddelbart med prednisolon 100 mg x1 og cyklofosfamid 100 mg (1,5 mg/kg). Etter 24 timer kom det rask bedring av proksimal muskelstivhet, og etter fire uker var aktivert partiell tromboplastinttid normalisert og faktor VIII-nivå målt til over 80 %. Inhibitor mot faktor VIII kunne ikke lenger påvises. Ved kontroll fire måneder etter behandlingsstart var prednisolon trappet ned til 20 mg x1, cyklofosfamid ble gitt videre i uendret dose. Pasienten var i god allmenntilstand, og det var ingen nye kliniske momenter. Kort tid etter ble pasienten innlagt med akutt innsettende dyspné. CT thorax påviste lungeemboli. Faktor VIII ble målt til > 100 %, faktorinhibitor kunne ikke påvises og man valgte å behandle med lavmolekylært heparin. CT thorax og abdomen viste ingen malignitetssuspekte forandringer.
Behandling av ervervet hemofili er assosiert med trombose (9). Siden pasientene gjerne har høy alder og ofte komorbiditet, er det ikke mulig å si om ervervet hemofili i seg selv gir økt risiko for trombose.
Pasienten ønsket ikke ytterligere malignitetsutredning. Man valgte å avslutte behandling med cyklofosfamid. Prednisolon ble trappet ytterligere ned til 10 mg, og pasienten ble skrevet ut. To år senere er pasienten fortsatt i live. Det har siden ikke vært konstatert ondartet sykdom, tromboembolisk hendelse eller blødningsmanifestasjoner.
Diskusjon
Ervervet hemofili er en sjelden tilstand og skyldes autoantistoffer mot koagulasjonsfaktorer, hyppigst faktor VIII. Dette er autoantistoffer som enten hemmer den biologiske aktiviteten til faktor VIII eller fjerner faktor VIII fra sirkulasjonen. Hos pasienter med alvorlig medfødt hemofili A (< 1 % faktor VIII-nivå) vil om lag 30 % utvikle inhibitorer (10). Utvikling av faktor VIII-inhibitorer hos pasienter uten medfødt hemofili A kalles ervervet hemofili. Tilstanden er svært sjelden, med insidens 1,3 – 1,5 per million per år (11, 12). Inhibitorer mot andre koagulasjonsfaktorer forekommer, men er enda sjeldnere. I perioden 2003 – 08 ble alle nydiagnostiserte pasienter med ervervet hemofili i en rekke europeiske land registrert i det europeiske registeret for ervervet hemofili (European Acquired Haemophilia Registry, EACH2) (6). Median debutalder var 73,9 år, og 75 % av pasientene var over 61,4 år. Fordelingen menn: kvinner var 1 : 0,88 (6). Ettersom kvinner lever lenger enn menn, er muligens ervervet hemofili hyppigst hos menn for personer over 65 år, mens for personer under 65 år er tilstanden noe hyppigere hos kvinner. Denne forskjellen utgjøres av kvinner med ervervet hemofili i postpartumperioden. Data for samtlige pasienter behandlet ved Rikshospitalet i perioden 1997 – 2004 er publisert (13). Materialet inkluderte 14 pasienter (åtte kvinner og seks menn), med median alder 78 år (58 – 94) og beregnet insidens 0,5 tilfeller per million. Dette er antakelig et underestimat, da tilstanden lett overses hos eldre med komorbiditet, i tillegg til at ikke alle pasienter får behandling ved Rikshospitalet. Hos fire av pasientene ble det funnet en lidelse med kjent assosiasjon til ervervet hemofili.
Ervervet hemofili er sjelden hos personer under 50 år, og kvinner i postpartumperioden utgjør flesteparten av disse pasientene (6). Tilstanden er sjelden manifest ved fødselen. Faktor VIII-inhibitorer utvikler seg som regel en tid ut i postpartumperioden, med median debuttidspunkt om lag tre måneder etter fødsel (6). Faktor VIII-inhibitor kan ses så sent som 12 måneder etter fødselen.
Isolert spontan forlenget aktivert partiell tromboplastinttid hos personer med blødninger gir mistanke om medfødt eller ervervet faktormangel. Normal aktivert partiell tromboplastinttid utelukker tilstanden. Ved forlenget aktivert partiell tromboplastinttid og mistanke om ervervet hemofili må blodprøven sendes til spesiallaboratorium for måling av faktor VIII-nivå og inhibitortiter. For raskt å sannsynliggjøre tilstedeværelse av inhibitorer kan man gjøre en såkalt «blandetest» (5). Ved denne testen blander man plasma fra pasienten med plasma fra en frisk person (normalplasma). Dette oppbevares så ved 37 °C i to timer. Deretter analyseres aktivert partiell tromboplastinttid i pasient-normalplasmablanding og i normalplasma. I løpet av inkuberingstiden vil det være spontant henfall av koagulasjonsfaktorer. Dette fører til at aktivert partiell tromboplastinttid i begge prøvene forlenges med ca. 4 – 6 sekunder. Hvis differansen mellom aktivert partiell tromboplastinttid i pasient-normalplasmablanding og normalplasma er over 10 sekunder, foreligger det en inhibitor som er i stand til å nøytralisere tilført faktor fra normalplasma. Lupusantikoagulans vil i blandetest føre til vedvarende forlenget aktivert partiell tromboplastinttid, men da vil den kliniske problemstillingen være økt risiko for trombose.
Vanligste debutsymptomer ved ervervet hemofili er subkutane og retroperitonale blødninger fulgt av gastrointestinale, lunge-, urogenitiale og cerebrale blødninger. Som hos vår pasient kan man ofte se betydelige blødninger etter venepunksjoner og subkutan blødning etter blodtrykksmåling. Dette skiller seg fra medfødt hemofili, der vanligste blødninger er traumatiske muskel- eller leddblødninger. Det er angitt at ved ervervet hemofili er ca. 10 – 20 % av blødningene fatale, derav 2 % intracerebrale (14). 85 % av pasientene opplever livstruende blødninger (14). På det tidspunktet vår pasient ble vurdert for fasciotomi ble ikke muligheten for ervervet hemofili vurdert. En operasjon på dette tidspunktet ville medført store og sannsynligvis fatale blødninger.
Nivå av inhibitor/autoantistoff måles i Bethesda-enheter (BE). Inhibitor med nivå 1 BE betyr at denne pasientprøven reduserer/nøytraliserer målt faktor VIII-nivå med 50 % hvis den blandes med lik mengde plasma fra en frisk person. Hos pasienter med faktor VIII-inhibitor er det ikke alltid samsvar (dvs. en klar invers korrelasjon) mellom nivå av inhibitor og nivå av målt faktor VIII. Selv om pasientene skulle ha faktor VIII-nivå svarende til et område som gir liten risiko for blødning hos pasienter med medfødt faktor VIII-mangel, ser man en betydelig større blødningstendens. Fatale blødninger kan også ses flere måneder ut i forløpet. Denne risikoen vedvarer så lenge inhibitorer er til stede. Eradikasjonsbehandling med sikte på å fjerne inhibitorer bør derfor startes tidligst mulig.
Noen data tyder på at inntil 25 % av pasientene med ervervet hemofili vil oppleve spontan remisjon (15). Det finnes imidlertid ingen måte å forutse hvilke pasienter som oppnår remisjon uten immunsuppressiv behandling. Man skal derfor raskest mulig etter bekreftet diagnose starte behandling med sikte på å eradikere faktor VIII-inhibitoren. Standard behandling er enten steroider alene eller steroider kombinert med cyklofosfamid. Behandlingsregime angitt i de nordiske retningslinjene er prednisolon 1mg/kg i seks uker for så raskt å trappe ned, kombinert med cyklofosfamid 1,5 – 2 mg/kg daglig med behandlingslengde 3 – 4 måneder (5). For cyklofosfamid finnes det også andre perorale og intravenøse doseringsalternativer. I publiserte data fra det europeiske observasjonsregisteret for ervervet faktor VIII-mangel ble langvarig remisjonsrate for behandling med steroider alene angitt til å være 48 %, og for steroider kombinert med cyklofosfamid 70 % (11). Behandlingsrespons er uavhengig av underliggende tilstand, men synes å være påvirket av faktor VIII-nivå og inhibitortiter ved diagnosetidspunktet. Median tid til residiv ser ut til å være uavhengig av behandlingtype og er 4 – 5 måneder etter avsluttet behandling (11).
Konklusjon
Ervervet hemofili er en sjelden tilstand med autoantistoffer mot koagulasjonsfaktorer, hyppigst faktor VIII. Tilstanden rammer hovedsakelig eldre av begge kjønn og en sjelden gang yngre kvinner i postpartumperioden. Fordi dette er en sjelden tilstand og de fleste pasientene er eldre med komorbiditet, er tilstanden lett å overse. Blødning behandles med faktor VIII-konsentrat dersom inhibitortiteret er lavt (< 5 BE), ellers benyttes aktivert protrombinkompleks-konsentrat eller aktivert faktor VIIa. Alvorlig blødning kan forekomme ved faktor VIII-nivåer, som ellers er ansett å innebære liten blødningsrisiko. Pasienter med ervervet hemofili og blødninger bør håndteres ved sykehus med tilstrekkelig tilgang på faktorkonsentrat og hematolog med spesialkompetanse innen koagulasjonsforstyrrelser. Behandling tar sikte på å eradikere faktor VIII-inhibitorer og bør startes snarest mulig. Steroider kombinert med cyklofosfamid gir best vedvarende remisjon. Pasienter med ervervet hemofili har muligens økt risiko for tromboembolisk hendelse.
