En kvinne i 40-årene ble innlagt ved universitetssykehus med to ukers sykehistorie med økende hodepine, gangvansker og nedsatt allmenntilstand. Hun hadde mange års sykehistorie med uveitter og nevrologiske symptomer bak seg, men på tross av omfattende utredning var diagnosen fortsatt usikker.
En kvinne i 40-årene ble innlagt fra lokalsykehuset for utredning av økende hodepine, gangvansker og nedsatt allmenntilstand. Symptomene hadde forverret seg de siste to ukene, men hun hadde en lang sykehistorie bak seg. I forbindelse med utredningen ble pasientens tidligere sykehistorie gjennomgått.
Allerede for 15 år tilbake hadde pasienten blitt penicillinbehandlet grunnet flåttbitt og erythema migrans. Få år etter dette, rundt ti år før den aktuelle innleggelsen, fikk hun bilateral panuveitt. Det var positive funn av IgM- og IgG-antistoffer mot Borrelia i serum, samt IgG i spinalvæsken, og hun var blitt behandlet med ceftriakson intravenøst.
Den bilaterale panuveitten ble den gangen oppfattet som disseminert Lyme-borreliose. Uveitt er et av mange funn som kan forekomme både i tidlig og sen fase av sykdommen (1).
Til tross for antibiotikabehandling hadde uveitten persistert. Man kunne da ikke utelukke autoimmun uveitt, og et halvt års tid etter første uveitt ble det startet med peroral kortikosteroid behandling. På grunn av dårlig klinisk respons la man til metotreksat, som etter hvert ble erstattet med ciklosporin. Etter ca. tre års behandling med ciklosporin hadde øyeforandringene bedret seg og medikamentet ble seponert. To måneder etter seponering fikk pasienten imidlertid økende hodepine og etter hvert feber. Hun ble innlagt på lokal nevrologisk avdeling, og derfra ble hun overflyttet til universitetssykehus for første gang.
Ved innkomst hadde hun da lett høyresidig hemiparese, venstresidig facialisparese, høyresidig ataksi, blikkretningsnystagmus mot venstre, høyresidig hyperrefleksi og plantarinversjon. Senkningsreaksjonen (SR) var lett forhøyet 19 mm/h (< 17 mm/h), C-reaktivt protein var normalt (< 5), spinalvæske inneholdt 159 · 10⁶/l celler (62 mononukleære, 97 polymorfonuklære) (< 5 · 10⁶/l), protein var forhøyet 0,95 g/l (< 0,5 g/l) og glukose 2,8 mmol/l. Spinalglukose-blodglukose-ratio var 0,6 (> 0,5). Det var ingen patologiske funn ved IgG- og IgM-Borrelia-serologi eller mikrobiologiske undersøkelser av både serum og spinalvæske. Blodprøver viste grenseverdi for antinukleære antistoffer (ANA), men tester for antinøytrofile cytoplasmaautoantistoffer (ANCA) og Waalers reaksjon var negative.
MR caput viste forandringer i mesencephalon, øvre del av pons og venstre cerebellarpedunkel, og i høyre hippocampus. Hjerneforandringene ble tolket som iskemiske lesjoner (fig 1). MR-angiografi var uten patologiske funn. Derimot var EEG patologisk, med intermitterende langsom thetaaktivitet frontotemporalt bilateralt. Enfotonstomografi (SPECT) viste nedsatt perfusjon i høyre cerebellarhemisfære og basalt temporalt på høyre side.
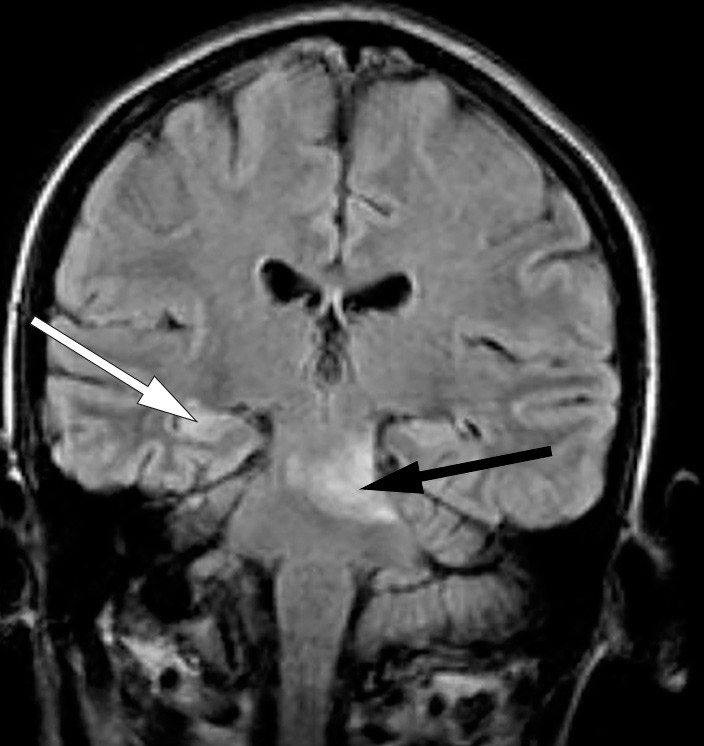
Figur 1 MR caput med koronal FLAIR (fluid attenuated inversion recovery) tatt ved første episode med hjerneaffeksjon som viser høysignalforandringer i pons og mesencephalon, mest uttalt på venstre side (svart pil), og i høyre hippocampus (hvit pil)
På bakgrunn av feber, forhøyet celletall i spinalvæsken og nevrologiske utfall hadde vi mistanke om uspesifikk viral eller bakteriell meningoencefalitt. Pasienten ble derfor behandlet både med antibiotika og antivirale midler, men behandlingen ble stoppet etter at mikrobiologiske prøver var negative. Dette inkluderte Borrelia-serologi både i serum og spinalvæske, noe som er ikke uvanlig etter vellykket behandling (2). Ved bakteriell meningoencefalitt er celletallet i spinalvæsken vanligvis sterkt forhøyet (gjerne over 1 000 hvite blodceller/mm³), glukoseratioen lav, og allmenntilstanden forverres raskt med feber og bevissthetsendring. Pasienten hadde imidlertid kun lett forhøyet celletall og normal glukoseratio. Bred utredning med tanke på autoimmun, inflammatorisk, infeksiøs og malign sykdom var uten positive resultater. Differensialdiagnostisk ble det vurdert om MR-funnet av iskemiske lesjoner kunne peke i retning av progressiv multifokal leukoencefalopati (PML), en demyeliniserende tilstand som oftest rammer immunsupprimerte pasienter grunnet reaktivering av JC-virus (3). Spinalvæskeundersøkelse hos vår pasient viste ikke funn av JC-virus, og derfor vurdert vi progressiv multifokal leukoencefalopati som lite sannsynlig. Tilstanden ble isteden oppfattet som et uveomeningealt syndrom. Dette er en inflammatorisk multisystemsykdom som affiserer øye, indre øre, hud og meninger (4).
Pasienten ble behandlet med intravenøs Solu-Medrol 1 g/d i fem dager, etterfulgt av peroral prednisolon 40 mg daglig. Ved tre måneders kontroll var pasienten i god klinisk bedring og cerebral MR viste også betydelig tilbakegang av signalforandringer. Prednisolondosen ble langsomt redusert, men senere ble ciklosporin gjenopptatt grunnet nye uveitter.
Etter seks-sju år med stabilt klinisk bilde, og ti år etter første uveitt, ble pasienten på nytt innlagt ved lokalsykehuset med økende hodepine, gangvansker og nedsatt allmenntilstand. Blodprøver viste forhøyet SR på 50 mm/h, men normale funn av C-reaktivt protein og leukocytter. I spinalvæsken var det økt celletall med 23 · 10⁶/l leukocytter, totalprotein 0,68 g/l, men normal glukoseratio. MR caput viste progresjon av forandringene med tilkommet signalavvik i mediale venstre temporallapp (amygdala, uncus og fremre hippocampus), i basalgangliene og subkortikalt i begge hemisfærer. Ny gjennomgang av laboratorieprøver ga ikke positivt holdepunkt for spesifikke virale eller bakterielle agens. Behandling med Solu-Medrol intravenøst ble startet, men pga. rask klinisk forverring med økende hodepine, gangvansker og nedsatt allmenntilstand ble hun innlagt ved universitetssykehus, slik det er beskrevet innledningsvis.
Det kliniske bildet, spinalvæskefunnet og forandringene påvist med MR pekte i retning av immunmediert tilstand, hvorav uveomeningealt syndrom ble ansett som mest sannsynlig. Det ble derfor startet med intravenøs Solu-Medrol som ofte er førstevalg ved inflammatorisk tilstand (5).
I løpet av et par dager ble pasienten somnolent og fikk økende muskeltonus i alle fire ekstremiteter, økte reflekser og bilateral plantarinversjon. CRP steg til 130 mg/l (< 5 mg/l), leukocytter til 18 · 10⁶/l (4,5 · 10⁶/l) og SR til over 100 mm/h. Gjentatte spinalpunksjoner viste lett leukocytose (16 · 10⁶/l celler) og lett forhøyet totalprotein (0,66 mg/l). Isoelektrisk fokusering av spinalvæske var uten tegn til intratekal IgG-produksjon. EEG endret seg på seks dager fra nærmest normalt til svært patologisk, med generalisert høyspent, langsom theta- og deltaaktivitet samt hyppige, skarpe potensialer frontotemporalt med trifasisk utseende. MR caput viste progresjon av signalavvikene i mediale venstre temporallapp (amygdala, uncus og fremre hippocampus) i basalgangliene og subkortikalt i begge hemisfærer (fig 2a og b).
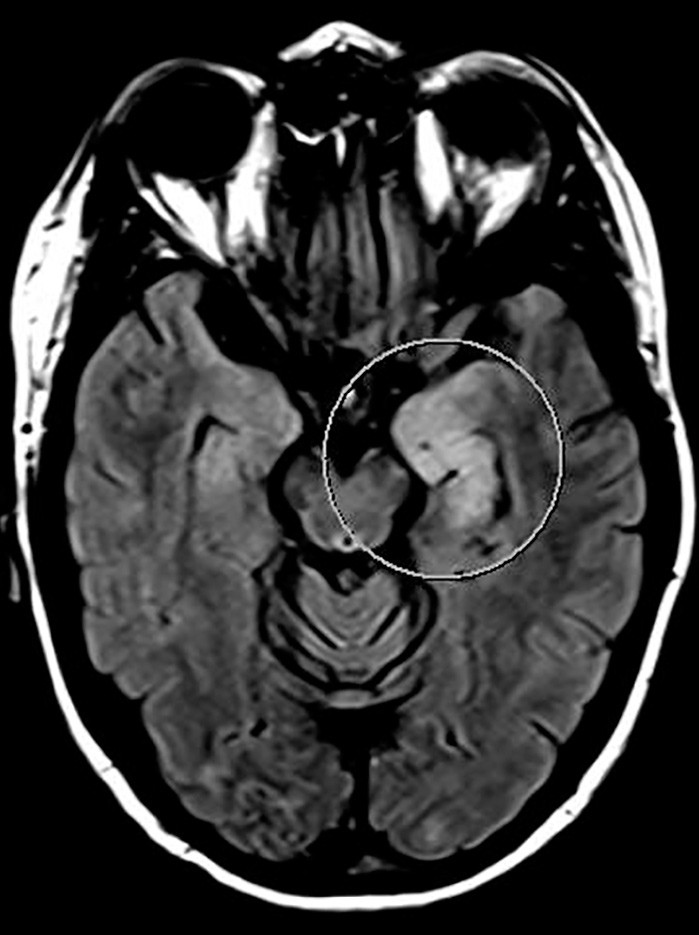
Figur 2a MR caput med aksial FLAIR tatt under den aktuelle episoden med hjerneaffeksjon, viser signalforandringer i venstre temporallapp, med affeksjon av amygdala, uncus og hippocampus (markert med sirkel)
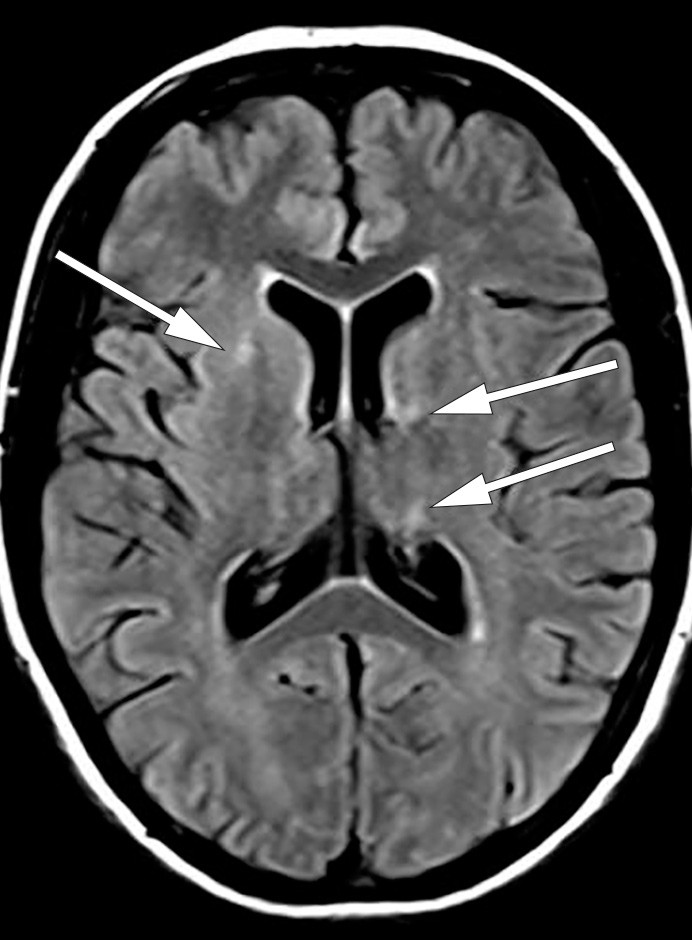
Figur 2b MR caput med aksial FLAIR viser høysignalforandringer i fremre bein av høyre capsula interna og i venstre caput nucleus caudatus og thalamus (pulvinar thalami)
Signalforandringene i disse områdene var uten tegn til diffusjonsrestriksjon, noe som talte mot ferskt iskemisk infarkt. MR-bildene ga derimot mistanke om viral encefalitt eller limbisk encefalitt. Det var imidlertid ikke påvist affeksjon av insula, basale frontallapp og gyrus cinguli, som ofte er involvert ved limbiske encefalitter (6).
Malignitetsutredning, inklusive undersøkelse av hodet og halvkropp med 18F-fluoro-2-deoksyglukose (FDG)-PET kombinert med CT (18-FDG-PET-CT) var også negativ. Det ble imidlertid påvist svakt positiv anti-NMDA-reseptorantistoff, og det ble startet intravenøs behandling med immunoglobuliner 0,4 mg/kg/d i fem dager.
Anti-N-metyl-D-aspartat (NMDA)-reseptorencefalitt er en av subtypene av limbisk encefalitt. Anti-NMDA-reseptorencefalitt er en dynamisk sykdom som utvikler seg i fem faser: psykose, epileptiske anfall, katatonisk tilstand, hyperkinetisk stadium og stadier med autonome forstyrrelser og pustevansker (7). Sykdommen rammer oftest barn (8) og unge voksne og kan være assosiert med svulster, ovarialt teratom og testistumor. Spinalvæskefunn viser typisk lett pleocytose og normalt eller lett forhøyet proteinnivå. Mer enn halvparten har også intratekal immunoglobulinproduksjon. Anti-NMDA-reseptorantistoff kan påvises både i serum og i spinalvæske. Pasienten vår hadde ikke slik klinisk utvikling, men på grunn av rask forverring og svakt positivt anti-NMDA-reseptorantistoff, valgte vi å starte med immunoglobuliner, som er førstevalg ved limbisk encefalitt (7).
Pasientens tilstand ble ytterligere forverret. Hun reagerte ikke på smertestimulus eller tilrop, ble spastisk i alle ekstremiteter, nakkestiv og hadde bilateral plantarinversjon. MR caput viste ytterligere progresjon av høysignalområder i begge hemisfærer, både i basalganglier, subkortikalt og kortikalt (fig 3). På grunn av manglende respons på steroider og immunoglobuliner samt rask og betydelig klinisk forverring, valgte vi å starte med immunsuppressiv behandling i form av rituximab 1 000 mg intravenøst.
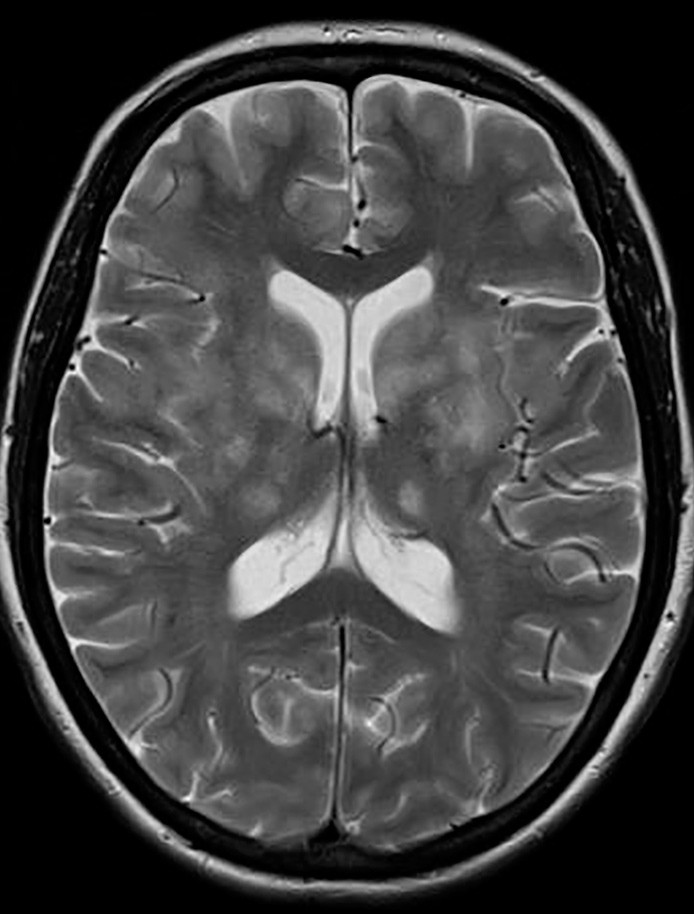
Figur 3 MR caput med aksial T2 viser progresjon av signalforandringer i basalganglier og thalamus bilateralt
Til tross for intensiv behandling skjedde en markant forverring i pasientens tilstand. Vi tolket tilstanden fortsatt som immunmediert og valgte derfor rituximab etter behandlingsprotokoll for revmatoid artritt.
Et par dager etter første infusjon var pasienten i klinisk bedring. Hun kunne kommunisere med korte setninger, ga blikkontakt og beveget armer og bein på oppfordring. Hennes kliniske tilstand ble imidlertid forverret rett før andre rituximabdose to uker senere. Pasienten ble sløv og trøtt, etter hvert ikke kontaktbar, hadde blikkdeviasjon mot venstre, bilateral plantarinversjon og feber. Undersøkelse av spinalvæske viste nå 409 · 10⁶/l celler (3/4 polynuklære) og totalprotein på 1,2 g/l. Nye MR-bilder av hjernen viste progresjon av forandringene i hvit substans, først og fremst i venstre hemisfære, men noe tilbakegang av signalforandringene subinsulært på venstre side.
Pasienten fikk andre rituximabdose og startet med prednisolon 30 mg daglig. En måned etter behandling kunne hun gå med støtte, hadde lett dysartri og apraksi. Hennes kliniske tilstand ble stadig bedre. MR caput utført to måneder etter andre rituximabinfusjon viste nærmest fullstendig tilbakegang av områdene med økt T2-signalintensitet i hvit substans rundt sideventrikler og i basalganglier.
Pasientens visus ble imidlertid betydelig svekket. Øyelege fant okkluderte blodkar både på arterie- og venesiden samt blødninger i fundus (fig 4).
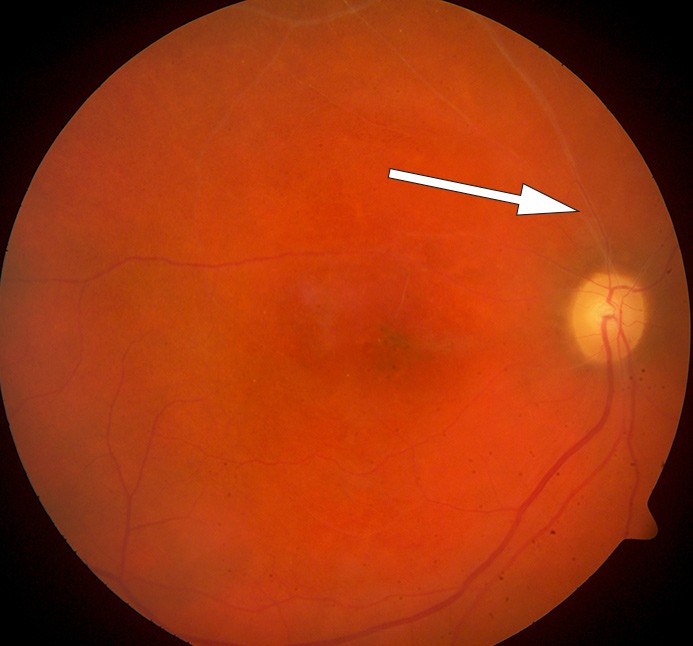
Figur 4 Fundusbilde av høyre øye. Okkluderte hvitlige kar både på arterie- og veneside
Slike vaskulittforandringer i øyebunnen ga mistanke om Behçets sykdom. Denne diagnosen var også vurdert tidlig i forløpet, men orale ulcerasjoner, som er vanlig ved denne tilstanden, var ikke observert under innleggelsene. Imidlertid kan potent immunsuppressiv behandling ha maskert slimhinnesår. På spørsmål om dette kom det frem at pasienten hadde vært plaget med residiverende orale ulcerasjoner i mange år. Tilstanden ble oppfattet som Behçets sykdom med affeksjon av øyne og sentralnervesystemet. Det var tilkommet en sikker bedring av de sentralnervøse manifestasjonene, men på grunn av økende vaskulittforandringer i øyebunnen begynte man tre måneder etter siste rituximabinfusjon med infliksimab 3 mg/kg etter behandlingsprotokoll for revmatoid artritt. Pasientens sykdomsbilde har holdt seg stabilt etter oppstart med infliksimab.
Diskusjon
Denne kasuistikken viser et utfordrende og sammensatt sykdomsbilde. Pasientens sykdomsforløp startet med uveitter som initialt ble oppfattet som borreliose. Det er imidlertid ikke uvanlig at pasienter med autoimmun sykdom også har forhøyede verdier av andre antistoffer, som skyldes kryssreaksjon (9). Sykehistorien illustrerer hvordan positive laboratorieprøver og behandling med immunsuppressive midler kan vanskeliggjøre diagnostikken. Allerede under første innleggelse på nevrologisk avdeling mistenkte man uveomeningealt syndrom. Ved uveomeningealt syndrom er det affeksjon av både uvea, retina og meningene. Etiologien er infeksiøs, inflammatorisk eller paraneoplastisk (10). Til dette syndromet hører tilstander som Behçets sykdom, granulomatøs polyangitt (Wegeners granulomatose), Vogt-Koyanagi-Haradas sykdom og akutt posterior multifokal plakoid pigment epiteliopati.
Behçets sykdom forekommer sjelden hos etnisk norske pasienter. Sykdommen rammer oftest befolkninger langs den historiske silkeveien i Asia og rundt Middelhavet (5, 11, 12). Så langt er det ikke etablert spesifikke blodprøver eller markører for å stille diagnosen på klinisk grunnlag. Anamnesen står derfor sentralt i utredningen (13). Behçets sykdom er en vaskulittsykdom som kan affisere kar av alle størrelser både på den arterielle og venøse siden av sirkulasjonen. Sykdommen har anfallsvise forløp, der vaskulitt og inflammatoriske forandringer kan forventes i nærmest alle typer vev (14). En internasjonal interessegruppe publiserte i 1990 klassifikasjonskriterier som fortsatt er i bruk (15). Det kreves tilstedeværelse av tre utbrudd med orale ulcerasjoner i løpet av 12 måneder og i tillegg minst to av følgende funn: tilbakevendende genitale ulcerasjoner eller arr, fremre eller bakre uveitter, retinale vaskulitter, papulopustulare hudlesjoner, erythema nodosum og positiv prikktest (patergi). Behçets sykdom kan også affisere andre organer som mage-tarm, lunge, ledd, hjerte-kar og sentralnervesystemet (5, 12).
De største publiserte studiene viser at 10 – 13 % av Behçet-pasientene har symptomer fra nervesystemet (5). Prevalensen er 2 – 50 % og er avhengig av region, etnisitet og kvaliteten på oppfølgingen (11). I aldersgruppen 20 – 40 år opptrer nevrologisk Behçets sykdom tre ganger hyppigere hos menn enn kvinner (5). De nevrologiske symptomene debuterer som regel ca. 3 – 6 år etter første tegn på systemisk Behçets sykdom. Affeksjon av nervesystemet viser to former, parenkymatøs eller vaskulær. Den parenkymatøse formen er typisk lokalisert til hjernestammen, som også var tilfellet hos vår pasient (5, 12, 13). Opptil 80 % av pasienter med nevrologisk Behçets sykdom har denne formen (13). Den vaskulære formen kjennetegnes med sinusvenetrombose, arterieokklusjoner og intrakraniale aneurismer (5, 13). Hodepine er det mest vanlige symptomet, og ca. tre av fire med affeksjon av hjernen debuterer med subakutt meningoencefalitt. Parese observeres i halvparten av tilfellene, en tredel har cerebellære utfall og like mange får dysartri. Transvers myelitt forekommer hos 10 – 30 %. Det perifere nervesystemet er sjelden affisert (5, 12, 16).
Det er heller ikke uvanlig med samtidig parenkymaffeksjon og trombose, oftest i sinus sagittalis. Prognosen ved sinusvenetrombose er som regel god (5, 12, 13). Mekanismen bak slike tromboser er ikke helt forstått. En mulig teori er epitelcelleaktivering som følge av inflammasjon (17).
Som nevnt finnes det ikke spesifikke diagnostiske prøver for Behçets sykdom. Diagnosen må derfor stilles på bakgrunn av kliniske manifestasjoner der slimhinneulcerasjoner forekommer hos samtlige pasienter (5, 15). Hos ca. 60 – 70 % av tyrkiske og japanske pasienter med sykdommen finner man positiv HLA-B51 i serum (18,19). Prøven er positiv hos bare 10 – 25 % av europeiske pasienter. Spinalvæskeundersøkelse avdekker forhøyede proteiner og pleocytose, isoelektrisk fokusering er ofte negativ (5). Det er også rapportert om pasienter med kun forhøyet totalproteinnivå i spinalvæsken (11). Patergitest viser papulopustulært erytem 24 – 48 timer etter intrakutant stikk med en steril 21G-nål på underarmen. Denne testen er positiv hos halvparten av pasientene fra endemiske områder, men bare 10 – 20 % av pasientene fra Nord-Europa får positive funn ved patergitest (20). Ved den parenkymatøse formen av nevrologisk Behçets sykdom viser MR caput med T2-vektede bilder typisk hyperintense lesjoner i hjernestammen, spesielt mesencephalon, og i basalganglier (5, 14). Forandringer i hvit substans subkortikalt, frontalt og temporalt kan også forekomme (5). Lesjonene er ofte kontrastoppladende (14, 21).
Pasienter med myelitt har på T2-vektede MR-bilder hyperintense lesjoner som ofte strekker seg over 2 – 3 virvelhøyder. Disse kan likne på demyeliniserende forandringer. CT-angiografi av pre- og intracerebrale kar og konvensjonell angiografi viser fravær av typiske vaskulittforandringer (5).
Histopatologiske prøver viser multifokal perivaskulær inflammasjon og nekrotiserende lesjoner samt lymfocyttinfiltrasjon (12, 21).
Det er ingen konsensus om den terapeutiske tilnærmingen til Behçets sykdom. En ekspertgruppe bestående av revmatologer, oftalmologer, indremedisinere, nevrologer og dermatologer har kommet med anbefalte retningslinjer for behandling (16). Som andre systemiske inflammatoriske sykdommer behandles pasienter ofte med steroider. Ved nye attakker eller manglende respons bør det ifølge disse retningslinjene legges opp til annen immunsuppressiv behandling. Avhengig av den kliniske tilstanden kan man velge å starte opp med kolkisin, metotreksat, azatioprin, TNF-alfaantagonister eller alfainterferon. Ciklosporin, som ofte er brukt i behandling av uveitt, regnes som kontraindisert ved nevrologisk Behçets sykdom. Studier har vist en høyere forekomst av denne tilstanden blant pasienter som behandles med ciklosporin (22). Om ciklosporin var medvirkende til utvikling av de nevrologiske manifestasjonene hos vår pasient, kan teoretisk ha vært mulig, selv om man ikke kan kalle det en utløsende årsak. Flere nye publikasjoner viser gode resultater av infliximab og rituximab ved Behçets sykdom (16, 23, 24). Erfaringer med disse medikamentene er få, og det er nødvendig med ytterligere studier og lengre observasjon. Sinusvenetrombose behandles med antikoagulasjon. Flere publikasjoner rapporterer også om gunstig tilleggseffekt av immunsuppressiver for antikoagulasjon av Behçet-pasienter med sinusvenetrombose (25).
Oppsummering
Behçets sykdom er sjelden hos etnisk norske og forekommer hyppigere hos personer med midtøstlig og asiatisk opprinnelse (19). Sykdommen bør likevel tas i betraktning ved uavklarte inflammatoriske tilstander. Tidlig diagnose er viktig for å unngå alvorlige irreversible skader. Denne kasuistikken viser betydningen av en nøyaktig gjennomgang av tidligere anamnese ved uavklarte subakutte encefalitter.
