En mann i 30-årene med vitiligo fra ungdomsårene fikk diagnosen diabetes mellitus type 1. Blodsukkeret ble etter hvert vanskelig å kontrollere, og man oppdaget at det ikke bare var bukspyttkjertelen som sviktet.
Pasienten fikk påvist glukose i urinen ved en rutinekontroll hos fastlegen da han var i starten av 30-årene. Han ble henvist videre til privatpraktiserende spesialist i indremedisin, som tolket tilstanden som type 2-diabetes. Pasienten ble behandlet etter datidens retningslinjer og startet med diett og glibenklamid. Glibenklamidbehandlingen fungerte ikke tilfredsstillende, og to måneder etter diagnosen ble han henvist og innlagt i medisinsk avdeling på lokalsykehuset. Under innleggelsen fremkom det at han fra tidlig i tenårene hadde hatt økende pigmenttap og som barn var han plaget med obstipasjon og magesmerter uten at han hadde vært nærmere utredet for dette. Foruten uttalt pigmenttap var det tilnærmet normale funn ved klinisk organundersøkelse og det var ingen spesiell sykdomsopphopning i nærmeste familie. Det ble påvist autoantistoffer mot glutaminsyre-dekarboksylase (GAD) og insulin.
Vår pasient fikk diabetesdiagnosen i voksen alder, og man mistenkte derfor initialt type 2-diabetes. Da han ikke responderte adekvat på behandlingen, ble diabetes type 1 med sen debut vurdert som alternativ diagnose, verifisert med påvisning av autoantistoffer. Pasienten ble gitt insulinbehandling (insulin actrapid) og han ble videre fulgt poliklinisk med akseptable blodsukkermålinger. Pigmenttapet debuterte i 13-årsalderen med en pigmentfri flekk midt på brystkassen, for siden å spre seg til hals, ansikt og hender. En allmennpraktiserende lege stilte diagnosen vitiligo klinisk da han var 16 år gammel, og ved innleggelsen hadde han typiske vitiligoforandringer blant annet i ansikt og på hender. Både funn av vitiligo og normal vekt støttet type 1 diabetes-diagnosen. Han gikk til regelmessige kontroller i både primær- og sekundærhelsetjenesten.
Ti år etter at diabetesdiagnosen ble stilt, ble han innlagt akutt i sykehus med alvorlig hypoglykemi. Han fortalte da om økende problemer med lave blodsukkermålinger og følingssymptomer over flere uker, dette til tross for at insulinbehandling, matinntak og aktivitetsnivå var uendret. Det kom også frem at han helt fra begynnelsen av tenårene hadde vært plaget med leggkramper. Hans egen lege hadde ved et tilfelle påvist lav serum-kalsium og gitt kalsiumtilskudd. Kontroll av laboratorieprøver viste s-kalsium 1,95 mmol/l (2,20 – 2,55 mmol/l), ionisert s-kalsium 1,07 mmol/l (1,13 – 1,28 mmol/l), s-fosfat 1,6 mmol/l (menn < 50 år: 0,75 – 1,65 mmol/l) og parathyreoideahormon (PTH) 1,8 pmol/l (1,3 – 6,8 pmol/l).
Laboratorieverdiene ble tolket til å være forenlige med primær hypoparatyreoidisme, og det ble startet vitamin D-behandling med alfakalsidol samt kontinuert kalsiumtilskudd. Pasienten presenterte nå både diabetes mellitus type 1, primær hypoparatyreoidisme og vitiligo. Vitiligo er autoimmunt betinget og rammer melanocytter. Pasienter med vitiligo har kjent høyere forekomst av autoimmun sykdom sammenliknet med normalpopulasjonen, som diabetes mellitus type 1 (ses hos 10 % av pasienter med vitiligo), autoimmun tyreoiditt (30 %), autoimmun gastritt (15 %) og pernisiøs anemi (5 %) (1 – 4). Pasienten hadde nå en autoimmun polyglandulær svikt, og den økte insulinfølsomheten gjorde at man også mistenkte hypokortisolisme.
Målinger av kortisol viste verdier i nedre normalområdet ved morgenprøve 273 nmol/l (normalt 120 – 600 nmol/l) og kveldsprøve 117 nmol/l (60 – 300 nmol/l), og samtidig adrenokortikotropt hormon (ACTH) på henholdsvis 24,6 pmol/l (2,0 – 11,6 pmol/l) og 7,3 pmol/l (< 4,6 pmol/l). Det ble funnet normale nivåer av aldosteron og renin. En poliklinisk synachtentest ga kortisolstigning fra 337 til 418 nmol/l (> 550 nmol/l).
Ved synachtentesten gis 0,25 mg av et biologisk aktivt peptid av adrenokortikotropt hormon (synacthen), noe som stimulerer binyrebarkens kortisolproduksjon maksimalt. Kortisol måles i serum før, og 30 og 60 minutter etter injeksjon. Ved normal respons stiger kortisol til over 550 nmol/l. I dette tilfellet tolket man resultatene til å være forenlig med lett primær binyrebarksvikt, og pasienten fikk forskrevet kortisonacetattabletter. Konstellasjonen binyrebarksvikt og hypoparatyreoidisme gjorde at man nå mistenkte at pasienten hadde autoimmunt polyendokrint syndrom type 1 (APS-1). I den videre oppfølgingen i spesialisthelsetjenesten ble man derfor spesielt oppmerksom på kliniske manifestasjoner som kan inngå i denne sykdommen (fig 1).
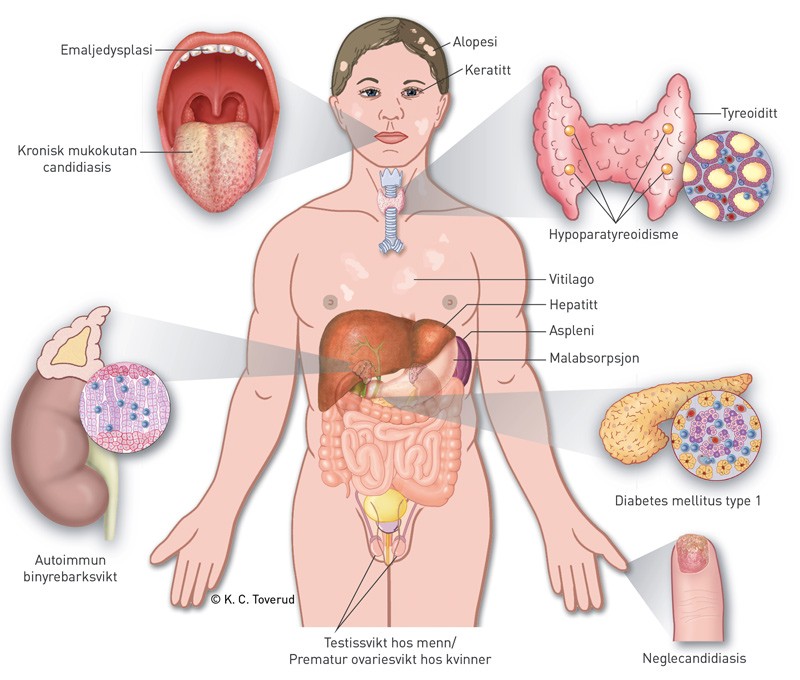
Figur 1 Kliniske manifestasjoner som kan forekomme hos pasienter med autoimmunt polyendokrint syndrom type 1 (APS-1)
Ny familieanamnese avdekket at hans yngre søster hadde fått primær binyrebarksvikt da hun var i 20-årene, noe som styrket mistanken om APS-1 som har autosomalt recessiv arvegang. Det ble påvist autoantistoffer mot proteinene aromatisk L-aminosyre-dekarboksylase (AADC), glutaminsyredekarboksylase (GAD), tryptofan hydroksylase type I/II (TPH I/II) og NACHT leucine-rich-repeat-protein 5 (NALP5), alle typiske for APS-1. Videre viste sekvensering av det autoimmune regulatorgenet AIRE homozygositet for en spleisesetemutasjon (c.879+1G>A/c.879+1G>A).
Mutasjonen ble vurdert å være patogen, og det passet også med at pasientens foreldre viste seg å være femmenninger.
Fem år senere (omkring 15 år etter diabetesdiagnosen) ble han plaget med analkløe. Klinisk undersøkelse ga mistanke om candidainfeksjon i genital- og analområdet, uten at man klarte å verifisere dette ved mikrobiologisk dyrkingsprøve. Ytterligere klinisk undersøkelse ved poliklinisk oppfølging i spesialisthelsetjenesten ga mistanke om kronisk soppinfeksjon i munnhule og hud/neglesenger. Kronisk candidose på hud, slimhinner og negler er én av hovedkomponentene ved APS-1.
Pasienten ble behandlet med flukonazol med tilfredsstillende effekt. Undersøkelse av tannlege/kjevekirurg bekreftet funn av candidainfeksjon i dyrkingsprøve og i tillegg emaljehypoplasi, som også er vanlig ved APS-1.
Diskusjon
Over en tiårsperiode ble vår pasient diagnostisert med flere autoimmune sykdommer; initialt vitiligo, etterfulgt av diabetes mellitus type 1, primær hypoparatyreoidisme og subklinisk primær binyrebarksvikt. Pasienten hadde i så måte en rekke av de kliniske manifestasjonene som inngår i APS-1 (fig 1).
Autoimmune polyendokrine syndromer (APS) er sjeldne sykdommer karakterisert ved autoimmun svikt i minst to endokrine kjertler (5). Basert på kliniske kriterier ble det i 1980 foreslått en klassifisering i tre hovedgrupper, som vist i tabell 1 (6 – 9). Polyendokrin svikt som faller utenfor disse tre hovedgruppene, omtales av enkelte som APS-4 (6).
|
Tabell 1 Oversikt over de ulike autoimmune polyendokrine syndromene (6 – 9)
|
|
Autoimmunt polyendokrint syndrom type 1 (APS-1)
|
Autoimmunt polyendokrint syndrom type 2 (APS-2)
|
Autoimmunt polyendokrint syndrom type 3 (APS-3)
|
|
Diagnostiske hovedkriterier
|
Kronisk mukokutan candidiasis
Hypoparatyreoidisme
Autoimmun binyrebarksvikt
(minst to av tre manifestasjoner)
|
Autoimmun binyrebarksvikt
og autoimmun thyreoideasykdom
og/eller diabetes mellitus type 1
|
Autoimmun thyreoideasykdom assosiert med annen autoimmun sykdom
(unntatt binyrebarksvikt, hypoparatyreoidisme, kronisk candida)
|
|
Sykdomsdebut
|
Barndom
|
Ungdom/voksen
|
Ungdom/voksen
|
|
Gener og arv
|
AIRE (autoimmunt regulatorgen), autosomalt recessivt
|
Humant leukocyttantigen (HLA) og andre, multifaktoriell
|
HLA og andre, multifaktoriell
|
|
Kjønnsfordeling
|
Kvinner = menn
|
Kvinner > menn
|
Kvinner > menn
|
|
Patogenese
|
Autoreaktive T-celler unnslipper negativ seleksjon og bidrar til vevsdestruksjon
|
Autoreaktive T- og B-celler bidrar til vevsdestruksjon
|
Autoreaktive T- og B-celler bidrar til vevsdestruksjon
|
|
Andre kliniske manifestasjoner
|
Alopesi
Vitiligo
Malabsorpsjon
Aspleni
Hepatitt
Emaljedysplasi
Keratitt
Tyreoiditt
Diabetes mellitus type 1
Testissvikt eller prematur ovariesvikt
|
Autoimmun gastritt
Cøliaki
Vitiligo
Alopesi
Testissvikt eller prematur ovariesvikt
|
Diabetes mellitus type 1
Atrofisk gastritt
Cøliaki
Vitiligo
Bindevevssykdom
Inflammatorisk tarmsykdom
|
|
Immunologisk fenotype
|
Autoantistoffer mot intracellulære enzymer, interferoner (interferon-ω og interferon-α) og Th17-cytokiner (IL17F og IL-22)
|
Autoantistoffer mot proteiner i affiserte organer: 21-hydroksylase (binyre), glutaminsyredekarboksylase, insulinomassosiert protein-2 (endokrin pancreas), anti-thyreoideaperoksidaseantistoff (anti-TPO) og thyreoideastimulerende hormon-reseptor-antistoff (TRAS)
|
Autoantistoffer mot affiserte organer: anti-TPO, TRAS (thyreoidea), intrinsisk faktor/parietalcelleantistoffer (ventrikkel), transglutaminase (tynntarm)
|
Autoimmunt polyendokrint syndrom type 1 (APS-1) er en sjelden monogen autosomalt recessiv sykdom. Tilstanden omtales også i litteraturen som autoimmun polyendokrinopati-candidiasis-ektodermal dystrofi (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, APECED). Den kliniske diagnosen stilles dersom pasienten har to av de tre hovedmanifestasjonene hypoparatyreoidisme, primær binyrebarksvikt og kronisk mukokutan candidose (10). Hvis et søsken har APS-1, er én hovedmanifestasjon tilstrekkelig (11). Det kliniske bildet er svært variabelt, da disse pasientene kan presentere en rekke andre autoimmune manifestasjoner (fig 1) (7, 8).
Siden syndromet opptrer sjelden og kan ha flere ulike kliniske manifestasjoner som debuterer med mange års mellomrom, kan det være vanskelig å diagnostisere, spesielt når pasienten debuterer med andre manifestasjoner enn hovedkriteriene. Det er funnet ulik forekomst i ulike befolkningsgrupper: 1: 90 000 i Norge (12), 1:130 000 i Irland (13), mens noen befolkningsgrupper har høyere forekomst, som 1:14 000 på Sardinia (14), 1: 25 000 i Finland (14) og 1: 9 000 blant persiske jøder (14).
Sykdommen debuterer vanligvis i barnealder, og ofte opptrer de tre diagnostiske hovedkriteriene i kronologisk rekkefølge: først candidainfeksjon, vanligvis i løpet av de første fem leveårene, deretter hypoparatyreoidisme før tiårsalderen og så binyrebarksvikt før fylte 15 år (7, 14). Mange pasienter fyller altså de diagnostiske kriteriene før fylte 20 år. Dersom sykdommen debuterer tidlig, er det større sannsynlighet for å bli rammet av et større antall kliniske manifestasjoner. Imidlertid finnes det også eksempler der pasienter presenterer ulike kliniske manifestasjoner uten å fylle de diagnostiske kriteriene, og det kan dermed gå lang tid fra første kliniske manifestasjon til endelig diagnose kan stilles, som illustrert i denne kasuistikken. At pasienten beskrevet her først presenterte sikker autoimmun sykdom i voksen alder, bryter med hva som er beskrevet å være normalt forløp av sykdommen. Pasienter med diabetes mellitus type 1 har imidlertid en generelt økt risiko for annen autoimmun sykdom, vanligst thyreoideasykdommer som forekommer blant 15 – 30 % av pasienter med diabetes type 1, 5 – 10 % diagnostiseres med atrofisk gastritt/pernisiøs anemi, 4 – 9 % har cøliaki, hos 0,5 % påvises Addisons sykdom og 2 – 10 % får vitiligo (15). Diabetes mellitus type 1 kan også inngå i autoimmune polyglandulære syndromer (8) (tab 1). Dette, samt familieanamnese omkring autoimmune manifestasjoner og autoimmun sykdom, bør vektlegges i oppfølgingen av diabetespasienter. Hos vår pasient viste det seg for eksempel senere at hans søster med Addisons sykdom også hadde APS-1, noe som ble bekreftet ved gendiagnostikk.
APS-1 skyldes mutasjoner i det autoimmune regulatorgenet AIRE (16). AIRE uttrykkes i medullære epiteliale celler i thymus. Her sørger det for at vevsspesifikke proteiner som insulin og C-reaktivt protein uttrykkes i små mengder, slik at umodne T-celler blir eksponert for disse. T-celler som reagerer på eget protein vil dermed kunne fjernes fra det immunologiske repertoaret ved negativ seleksjon. Om AIRE ikke fungerer, lekker autoreaktive T-celler ut i sirkulasjonen der de senere kan aktiveres og utøve organskade. Det er påvist omkring 100 ulike mutasjoner i AIRE-genet (17). Den vanligste i Norge, Storbritannia og USA er en 13bp-delesjon i ekson 8 (c.967_979del13) (12), mens i Finland og Øst-Europa dominerer den «finske» mutasjonen c.769C>T (p.R257X) (16). Det er imidlertid ikke funnet korrelasjon mellom de ulike mutasjonene og spesifikke fenotyper.
Den eksakte patogenesen ved APS-1 er fremdeles uklar. Man antar imidlertid at den autoimmune destruktive prosessen er T-cellemediert, men de fleste pasienter har også autoantistoffer mot autoantigen presentert i det affiserte vevet/organet. Autoantigenene man kjenner til er hovedsakelig organspesifikke enzymer (for eksempel 21-hydroksylase), interferoner og interleukiner. Enkelte autoantistoffer korrelerer til kliniske manifestasjoner, oftest med affeksjon i det organet autoantigenet uttrykkes. Autoantistoffenes rolle i den immunologiske reaksjonen er fremdeles uklar, men måling av autoantistoffer mot interferon-ω regnes nå å være en enkel diagnostisk test for APS-1 (18 – 21) fordi nesten alle pasientene har slike autoantistoffer. Positive autoantistoffer foreligger som regel før kliniske manifestasjoner gir seg til kjenne. I Norge tilbys analyse av autoantistoffer ved APS-1 ved Hormonlaboratoriet, Haukeland universitetssykehus.
Konklusjon
APS-1 er en sjelden, men trolig underdiagnostisert sykdom med et vidt spekter av kliniske manifestasjoner forårsaket av organspesifikk autoimmunitet. Da symptomene kan debutere med års mellomrom og er vanlige i befolkningen ellers, blir diagnosen ofte forsinket og mange ganger stilt først i voksen alder, selv om sykdommen debuterer i barneårene. Interferon-ω-antistoffer har stor diagnostisk treffsikkerhet og bør inngå i utredningen av disse pasientene. Interferonantistoffer bør måles hos barn og ungdom med en av hovedkomponentene ved APS-1, men bør også vurderes ved flere autoimmune manifestasjoner hos samme person, emaljehypoplasier, autoimmune hepatitter, uforklarlig malabsorpsjon og keratitter uten infeksiøs genese.
Behandlingen består i stor grad i å substituere de affiserte hormonaksene samt symptomatisk behandling av mer uspesifikke kliniske manifestasjoner. Mukokutan candidose behandles med antimykotika og bør følges tett for å unngå malignitetsutvikling, særlig i munnhulen. Det kreves oppmerksomhet rundt nyoppståtte kliniske manifestasjoner og forekomst av autoimmunitet i nær familie. Behandling og oppfølging bør være multidisplinær og ledes av en endokrinologisk avdeling.
