En mann i 70-årene ble henvist hematologisk poliklinikk grunnet redusert allmenntilstand og anemi. Blodprøver viste hemoglobin 9,7 g/dl (13,4–17,0), gjennomsnittlig cellevolum i de røde blodcellene (MCV) 115 fl (82–98), leukocytter 3,0 · 109/l (3,5–11,0), trombocytter 187 · 109/l (145–348), folat 7,2 nmol/l (> 8,0), kobalamin 693 pmol/l (175–700) og ferritin 831 µg/l (34–300). Tilstanden ble oppfattet som megaloblastisk anemi på grunn av folatmangel, og man startet med folatbehandling. Kontroll etter to måneder viste vedvarende anemi med hemoglobin 9,4 g/dl. Benmargsutstryk med May-Grünwald-Giemsa-farging viste en svært cellerik benmarg med en tydelig ekspandert erytropoese med megaloblastiske og dysplastiske trekk (bildet til venstre), mens ved jernfarging ble det funnet et uttalt antall ringsideroblaster (omtrent 30 % av cellene) (bildet til høyre).
Ringsideroblaster er atypiske, abnormale kjerneholdige erytroblaster (forstadier til modne røde blodceller) med granulering av jern sirkulært rundt kjernen (1). En ringsideroblast kjennetegnes ved at minst fem jerngranula omgir minst en tredel av kjernen. Anemien klassifiseres da som sideroblastisk anemi, som er en gruppe av både hereditære og ervervede tilstander (1). Vår pasient hadde refraktær anemi med ringsideroblaster. Det er en undergruppe av myelodysplastiske syndromer (MDS) som i hovedsak er karakterisert av anemi grunnet ineffektiv erytropoese (1). Disse syndromene deles i to: singel- og multilinjedysplasi. Vår pasient hadde singellinjedysplasi, siden kun erytropoesen var affisert. Ringsideroblaster forekommer også ved den beslektede tilstanden myelodysplastisk/myeloproliferativ neoplasi med ringsideroblaster og trombocytose (1). Denne tilstanden er karakterisert av anemi og trombocytose med store atypiske megakaryocytter i benmarg.
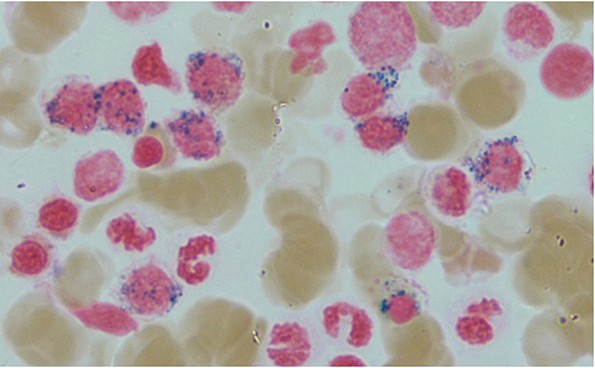
Ringsideroblastanemi kan være alt fra mikrocytær, som er det vanligste, til makrocytær, som hos vår pasient. Ved begge tilstander er ferritinverdiene forhøyet. Det er antatt at mitokondrier i erytroide forstadier er sentrale i patogenesen siden produksjon av hem skjer i mitokondriene. Opptil 80 % av pasientene har en mutasjon i genet SF3B1 som koder for et protein involvert i mitokondriefunksjonen (1, 2). Defekt i signalveier for jerntransport medfører at jern ikke blir omsatt normalt i mitokondriene. Opphopning av jern gir det karakteristiske morfologiske bildet med akkumulering av jern i mitokondrier rundt cellekjernen. Ved ringsideroblastanemi er det nå anbefalt å rekvirere mutasjonsanalyse (1, 3). SF3B1-genet var mutert hos vår pasient.
Myelodysplastiske syndromer er en gruppe maligne blodsykdommer karakterisert av klonal vekst i en eller flere hematopoetiske cellelinjer. Insidensen er om lag 4–5/100 000 per år og er økende ved alder (1, 3). Syndromene er svært forskjellige både i klinisk presentasjon, morfologisk bilde, genetisk profil og prognose. Sykdomsprogresjon med overgang til akutt myelogen leukemi forekommer. Behandlingen av myelodysplastiske syndromer har som mål å kontrollere symptomene fra de underliggende celledefektene og om mulig redusere sykdomsprogresjonen. Kurativ behandling med allogen stamcelletransplantasjon er forbeholdt yngre selekterte pasienter (3). Legemiddel som stimulerer erytropoesen kan bedre anemien. Hemoglobinnivået hos vår pasient steg etter behandling med erytropoietin (1, 3).
