Om lag 150 personer får amyotrofisk lateral sklerose (ALS) i Norge hvert år, og antallet er økende (1). Tradisjonelt har tilstanden blitt oppfattet som en rent motorisk sykdom karakterisert av progredierende pareser, men inntil halvparten utvikler også tegn til kognitiv svikt med atferds- og personlighetsforstyrrelser. En mindre andel utvikler frontotemporal demens (2). Pasientene dør vanligvis av respirasjonssvikt 2–3 år etter diagnosetidspunktet (3).
Amyotrofisk lateral sklerose deles gjerne inn i en sporadisk form uten andre kjente tilfeller i slekten (90–95 % av tilfellene) og en familiær form (5–10 % av tilfellene). Arvegangen ved sistnevnte er som oftest autosomalt dominant med ufullstendig og ofte ukjent penetrans. Siden oppdagelsen av mutasjoner i SOD1-genet i 1993 har det blitt påvist patogene mutasjoner i om lag 30 ulike gener ved familiær amyotrofisk lateral sklerose (3). Til sammen forklarer disse rundt 60–80 % av disse tilfellene (4). Slike mutasjoner finnes imidlertid også hos om lag 10 % av pasienter med sporadisk amyotrofisk lateral sklerose og hos betydelig flere som også har frontotemporal demens (5). Den vanligste årsaken til både arvelig amyotrofisk lateral sklerose og frontotemporal demens er økt antall repetisjoner av nukleotidene GGGGCC i C9orf72-genet (6). Testing av denne mutasjonen utføres ikke i Norge. Hyppigheten er derfor ikke kjent, men i andre land er den rapportert hos om lag 40 % ved familiær amyotrofisk lateral sklerose, 10 % ved sporadisk amyotrofisk lateral sklerose og 20 % ved frontotemporal demens (7, 8).
Det finnes per i dag ingen spesifikk terapi for noen av de genetiske variantene av amyotrofisk lateral sklerose. Ved den sporadiske formen er betydningen av et positivt funn for sykdomsrisiko hos slektninger usikker og kan utgjøre en stor tilleggsbelastning både for pasient og pårørende. Både den europeiske nevrologiforeningen (EFNS) og den europeiske interessegruppen for amyotrofisk lateral sklerose (ENCALS) frarådet derfor i 2004, 2007 og 2012 genetisk testing av pasienter med sporadisk variant, men åpnet for at testing kan utføres hos personer med familiær type eller atypiske kliniske funn der testing kan bidra til å avklare diagnosen (9–11). I Norge finnes det ikke offisielle faglige retningslinjer for utredning og oppfølging av amyotrofisk lateral sklerose. Anbefalinger i norsk elektronisk legemiddelhåndbok (Nevro-NEL) samsvarer med anbefalingene fra den europeiske nevrologiforeningen. Formålet med denne studien var å undersøke om praksis for genetisk utredning av pasienter med amyotrofisk lateral sklerose følger nasjonale og internasjonale anbefalinger.
Metode
Vi søkte elektronisk pasientjournal (EPJ) ved Akershus universitetssykehus for pasienter kodet G12.2 i henhold til ICD-10 (den internasjonale statistiske klassifikasjonen av sykdommer og beslektede helseproblemer) i perioden 31.12.2004–31.12.2014. G12.2 omfatter motornevronsykdommer, der amyotrofisk lateral sklerose er den desidert største pasientgruppen. Unntatt fra G12.2 er medfødte og arvelige varianter av spinale muskelatrofier. Vi vurderte hvorvidt diagnosen var korrekt samt om pasienten var primært utredet og fulgt ved Akershus universitetssykehus. Fra journalen innhentet vi så opplysninger om kjønn, alder, familieanamnese, genetisk testing og genetisk veiledning. Data ble oppbevart på sikker forskningsserver med avidentifiserte kliniske data separert fra koblingsnøkkelen.
Det er ingen klar konsensus for hva som skal defineres som familiær amyotrofisk lateral sklerose (12). I tråd med nyere anbefalinger basert på klinisk og etiologisk overlapp, ble pasienter med minst én første- eller annengradsslektning med amyotrofisk lateral sklerose eller frontotemporal demens klassifisert som familiær amyotrofisk lateral sklerose (13, 14).
Studien ble godkjent av personvernombudet ved Akershus universitetssykehus som en kvalitetsstudie. Det ble derfor ikke søkt godkjenning fra regional komité for medisinsk og helsefaglig forskningsetikk.
Resultater
Resultatene av journalgjennomgangen er vist i figur 1. Etter eksklusjon av ni feilkodede pasienter ble totalt 126 pasienter med amyotrofisk lateral sklerose vurdert ved Akershus universitetssykehus i studieperioden. Av disse ble 115 primært utredet og fulgt opp ved Akershus universitetssykehus, mens 11 pasienter med familiær type der det allerede var påvist ALS-assosierte mutasjoner, var henvist fra andre sykehus.
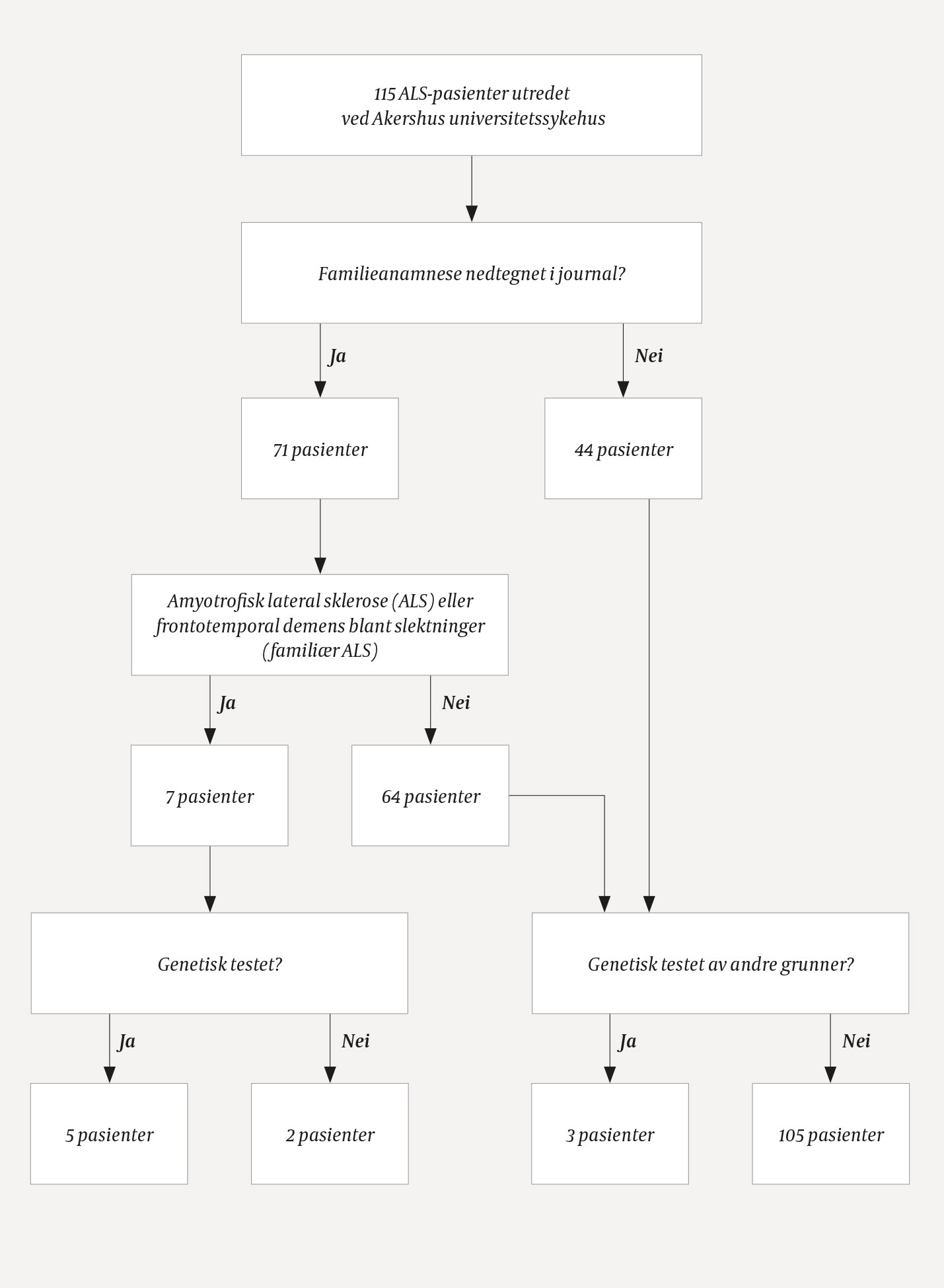
Figur 1 Genetisk utredning av pasienter med amyotrofisk lateral sklerose utredet ved Akershus universitetssykehus i perioden 2004–14
Ved gjennomgang av journalene fant vi at familieanamnesen var dokumentert hos 71 (62 %) av de 115 pasientene som hadde blitt utredet ved Akershus universitetssykehus. Av de 71 hadde syv (10 %) nære slektninger med amyotrofisk lateral sklerose eller frontotemporal demens forenlig med familiær amyotrofisk lateral sklerose. Blant disse ble det utført genetisk analyse hos fem pasienter. Det er ikke dokumentert i journalen hvorvidt genetisk utredning ble vurdert hos de øvrige to pasientene med familiær variant.
Det ble sendt blodprøve til diagnostisk genetisk analyse hos tre pasienter med sporadisk amyotrofisk lateral sklerose. Hos disse var undersøkelsen begrunnet med atypisk forløp, annen alvorlig nevrologisk sykdom (ikke motornevronsykdom eller frontototemporal demens) i nær slekt, eller den ble utført etter anmodning fra spesialist i medisinsk genetikk i forbindelse med pårørendes ønske om prediktiv testing.
Genetisk analyse ble nesten utelukkende utført på SOD1-genet, og hovedsakelig i siste halvdel av studieperioden. Med unntak av en pasient som fikk påvist heterozygot mutasjon uten sikker betydning, ble alle pasienter som fikk påvist ALS-relatert mutasjon tilbudt genetisk veiledning. I samråd med pasienten ble også pårørende informert og tilbudt genetisk veiledning. Ingen andre pasienter fikk tilbud om genetisk veiledning. Hos de to pasientene med antatt familiær amyotrofisk lateral sklerose som ikke fikk utført genetisk analyse, ble heller ikke genetisk veiledning nevnt i journalen.
Mot slutten av studieperioden fikk pasienter med påvist ALS-mutasjon tilbud om å delta i forskningsprosjekter. Fem donerte hudbiopsi for generering av induserbare pluripotente stamceller. Det ble også formidlet kontakt som muliggjorde deltagelse i en behandlingsstudie i utlandet.
Diskusjon
Våre funn viser en gjennomgående restriktiv praksis for genetisk utredning ved amyotrofisk lateral sklerose. Kun tre pasienter med sporadisk variant fikk utført diagnostisk gentesting, og dette var hos alle begrunnet i journalen. Med unntak av testing av én pasient med sporadisk variant initiert av spesialist i medisinsk genetikk ut fra pårørendes ønske om prediktiv testing, var vår praksis i tråd med europeiske anbefalinger som spesifikt har frarådet genetisk testing ved typisk sporadisk type (9–11).
To av syv pasienter med familiær variant fikk ikke tilbud om genetisk testing eller genetisk veiledning. Siden spørsmålet ikke var drøftet i journalen, kjenner vi ikke årsakene til at genetisk testing ikke ble utført. Fravær av eksplisitt begrunnelse kan gjenspeile lav bevissthet rundt betydningen av arvelighet, eller usikkerhet rundt tilgangen på genetisk testing og veiledning. En slik antagelse støttes av en studie fra 20 land som viser at nevrologer med stor erfaring med amyotrofisk lateral sklerose oftere tilbyr genetisk utredning enn mindre erfarne kolleger (14). En annen forklaring kan være et bevisst eller ubevisst ønske om å ikke bidra til å øke bekymringen for at slektninger også skal utvikle sykdommen. At familieanamnesen ikke var journalført hos nærmere 40 % av pasientene, er forenlig med begge disse forklaringene.
Den lave bruken av gentesting kan også gjenspeile et begrenset tilbud til slike analyser i Norge. En rekke ALS-relaterte gener, inkludert C9orf72 som er den klart hyppigste genetiske årsaken til amyotrofisk lateral sklerose, ble oppdaget i løpet av studieperioden. Oslo universitetssykehus analyserte imidlertid gjennom hele studieperioden kun SOD1, og analyserer fortsatt ikke andre ALS-relaterte gener. Mutasjoner i SOD1 forklarer kun en liten del av familiær amyotrofisk lateral sklerose (ca. 20 %), og analyse av kun dette genet regnes derfor ikke som tilstrekkelig utredning verken ved familiær variant eller atypiske forløpsformer av sporadisk type (14, 15). Den genetiske epidemiologien er ikke kartlagt i Norge, men ved å analysere C9orf72, FUS og TARDBP i tillegg til SOD1 vil man sannsynligvis kunne påvise den genetiske bakgrunnen til om lag to tredeler av de med familiær amyotrofisk lateral sklerose og 10 % av de med sporadisk type (14, 16).
Oppdagelsen av nye ALS-relaterte gener kan føre til at både pasienter, pårørende og leger oppfatter utredningen som mer meningsfull, ettersom den i større grad faktisk kan påvise eller utelukke genetisk årsak til sykdommen (17). Sykehuset i Telemark analyserer nå foruten SOD1 17 andre ALS-relaterte gener samt en rekke gener som er assosiert med andre motornevronsykdommer, men tester foreløpig ikke for C9orf72 (Helle Høyer, personlig meddelelse). Hovedargumentene for ikke å tilby genetisk testing ved sporadisk variant er at dette utfordrer hensynet til pårørende og pasientens rett til ikke å vite (15). Funn av en ALS-mutasjon hos en ALS-syk har vidtrekkende konsekvenser for friske slektninger. Disse må forholde seg til at deres risiko for å utvikle sykdommen kan være betydelig økt og må ta stilling til om de ønsker prediktiv testing, som etter norsk lov krever genetisk veiledning. Usikkerheten rundt betydningen av både positive og negative funn gjør at denne verken kan gi friske slektninger et tydelig beslutningsgrunnlag før testing eller sikker avklaring av egen risiko når testresultatet foreligger (18). Dette skiller seg fra Huntingtons sykdom, der en negativ gentest utelukker sykdommen og et positivt funn også gir viktig informasjon om forventet prognose (19).
Den relativt restriktive praksisen med å forbeholde genetisk testing til pasienter med familiær type eller atypisk forløp har i de senere år blitt utfordret (20), blant annet fordi den frarøver pasienten muligheten til å utforske årsaken til sykdommen (15). Dessuten gjør hyppige funn av ALS-mutasjoner hos pasienter med tilsynelatende sporadisk form grenseoppgangen mot familiær form mer arbitrær enn tidligere antatt (20, 21). Den relativt høye sannsynligheten for å oppdage en ALS-relatert mutasjon brukes således som argument både for en restriktiv og en liberal praksis for gentesting ved sporadisk amyotrofisk lateral sklerose (11, 17, 20). Økt innsikt i sykdommens genetiske årsaker er viktig for å forstå sykdomsmekanismene, og trolig vil mange fremtidige behandlingsstudier kreve molekylær utredning (22). Vår erfaring er at mange pasienter ønsker å delta i kliniske studier. Det er derfor sannsynlig at flere pasienter vil etterspørre molekylær utredning. Ettersom det vil bli mulig å teste for flere gener, øker også sannsynligheten for påvisning av genvarianter med usikker sykdomsassosiasjon og betydning for slektningers risiko. Da vil det også bli et betydelig større behov for genetisk veiledning.
Mange pasienter ønsker å vite årsaken til egen sykdom og risikoen for at den videreføres i slekten (18). I en spørreundersøkelse blant 5 591 amerikanske ALS-pasienter svarte 83 % av respondentene at genetisk testing burde tilbys alle pasienter med sykdommen, og 73 % mente at fordelene oppveiet ulempene (23). De som hadde gjennomgått genetisk testing, rapporterte overveiende positive erfaringer, uavhengig av sykdomsform (sporadisk eller familiær) og av hvorvidt de hadde fått genetisk veiledning (24). Responsraten i denne undersøkelsen var lav (8 %), og generaliserbarheten er derfor usikker. Diskrepansen mellom de positive tilbakemeldingene og den restriktive praksisen som gjenspeiles i våre funn og i europeiske anbefalinger, reiser likevel spørsmålet om nevrologer har en paternalistisk og overbeskyttende tilnærming, som kanskje også reflekterer eget ubehag ved å gå inn i disse spørsmålene.
Konklusjon
Vår studie avdekket en restriktiv bruk av gentesting ved amyotrofisk lateral sklerose. Med unntak av at familieanamnesen ikke var nedtegnet hos nesten 40 % av pasientene, var bruken i tråd med europeiske anbefalinger. Analysen omfattet ikke nyoppdagede og påvisbare ALS-assosierte gener som kunne ha økt den diagnostiske treffsikkerheten (17). Etablert genetisk diagnose kan åpne for deltagelse i behandlingsstudier og studier av sykdomsmekanismer (25), også ved mutasjoner i SOD1 som ser ut til å forekomme relativt hyppig i Norge (26). Flere pasienter med påvist mutasjon har donert hudbiopsi til generering av induserbare pluripotente stamceller og studier av sykdomsmekanismer, eller fått tilbud om å delta i behandlingsstudier i utlandet. Repertoaret av genetiske analyser bør derfor utvides i tråd med internasjonal standard.
