En mann i 70-årene ble utredet for sammensatte symptomer og sykdomsmanifestasjoner relatert til flere organsystemer. Plagene skulle vise seg å ha et felles utgangspunkt i en sjelden sykdom.
En tidligere sprek mann i 70-årene ble innlagt i akuttmottaket med forhøyet CRP, som hadde økt fra 41 mg/l til 90 mg/l (< 5) i løpet av tre dager, og forhøyet senkningsreaksjon på 51 mm/t (< 20). Ved klinisk undersøkelse hadde han et generalisert makuløst eksantem, som hadde debutert to måneder tidligere. Han hadde hatt ryggsmerter 2–3 uker i forkant av innleggelsen, men var i lett bedring.
To år før det aktuelle hadde pasienten blitt behandlet for en spondylodiskitt i lumbalcolumna, nivå L4/L5. Han fikk adekvat antibiotikabehandling i seks uker. Av øvrige sykdommer hadde han gjennomgått gjentatte abdominale operasjoner i etterkant av en lyskebrokkoperasjon 14 år tidligere. Han ble sist operert året før det aktuelle, da for midtlinjebrokk og subileus. Han brukte losartan for hypertensjon og fikk substitusjonsbehandling med folsyre, ellers brukte han ingen faste medikamenter.
På grunn av tidligere sykehistorie, nyoppståtte ryggsmerter og forhøyde infeksjonsmarkører var innleggende lege bekymret for en ny spondylodiskitt.
Supplerende blodprøver i akuttmottaket viste spontant forlenget protrombintid med INR på 1,4 (< 1,1) og et fall i CRP til 44 mg/l fra 90 mg/l. Det var utslag på urinstiks med 2+ for leukocytter, positiv nitritt og 2+ for protein.
Spondylodiskitt er en sjelden tilstand (1), og det var to år siden pasienten hadde blitt adekvat behandlet for dette. Han var afebril, og verken anamnese eller klinisk undersøkelse gav sikre holdepunkt for infeksjon. Det var ingen symptomer på perifere nevrologisk utfall og ingen kjente traumer. Smertene hadde blitt lindret av paracetamol, og residiv av spondylodiskitt ble vurdert som lite sannsynlig. Han hadde tegn til koagulopati, med spontant forhøyet INR, men samtidig normale albuminverdier, som talte imot leversyntesesvikt (2). Pasienten hadde nylig begynt med folsyrebehandling på grunn av biokjemisk bekreftet mangeltilstand, og differensialdiagnostisk kunne vitamin K-mangel knyttet til malabsorpsjon ha bidratt til koagulopati. Pasientens utslett var asymptomatisk og uendret siden debut.
Pasienten ble sendt fra akuttmottaket med diagnosen urinveisinfeksjon som ble behandlet med pivmecillinam 400 mg x 3 i fem dager. Det ble anbefalt videre oppfølging hos fastlege. To dager etter utskrivning forelå svar på urindyrkning, som ikke viste bakterievekst.
Urinveisinfeksjon er sjelden hos menn uten underliggende risikofaktorer som urinkateter, strukturelle avvik i urinveiene eller nevrologisk sykdom (3). Totalt sett var det ikke sterke holdepunkter for urinveisinfeksjon hos pasienten, og antibiotika burde sannsynligvis ikke ha blitt gitt.
To uker etter at pasienten kom hjem fra sykehuset, viste blodprøver tatt hos fastlegen nytilkommet anemi med hemoglobin 11,5 g/dl (13,4–17). Automatisk differensialtelling tatt ved fastlegekontoret anga «blastalarm», og det ble utført supplerende blodutstryk som ble vurdert av hematolog ved lokalsykehuset. Blodutstryket viste en lett venstreforskyvning med noen umodne forstadier av granulocytter, men det var ingen tegn til blaster.
Venstreforskyvning betegner påvisning av en større andel umodne granulocytter i blodbildet. Årsaken kan være reaktiv, som ved infeksjonssykdommer, men blodbildet kan også gi mistanke om hematologisk malignitet eller infiltrasjon i beinmarg av annen årsak (4).
Grunnet uklare funn i blodbildet ble pasienten henvist til hematologisk poliklinikk, og første konsultasjon fant sted én måned etter vurderingen i akuttmottaket. Ryggsmertene var da forsvunnet, men pasienten beskrev redusert allmenntilstand med tretthet og slapphet samt et vekttap på 3–4 kg de siste par månedene. Han fortalte også om ubehag som flatulens og løs avføring etter måltider. Ved klinisk undersøkelse var det normale funn utover det kjente asymptomatiske utslettet på truncus og proksimale ekstremiteter, som var en kombinasjon av brunlig makuløst og erytematøst eksantem. Supplerende blodprøver viste hemoglobin 10,5 g/dl og senkningsreaksjon 64 mm/t. I tillegg var det forhøyede leverprøver med gammaglutamyltransferase (γ-GT) 140 U/l (15–115), alkalisk fosfatase (ALP) 278 U/l (35–105), koagulopati med INR på 1,4 og aktivert partiell tromboplastintid (APTT) på 57 s (30–44). Proteinelektroforese i serum påviste en M-komponent, kvantitert til 2,7 g/l og klassifisert som immunoglobulin M med tilhørende frie lette kjeder type kappa.
Pasienten hadde symptomer fra flere organsystemer, mest uttalt i hud og gastrointestinalkanalen, og var preget av den reduserte allmenntilstanden og vekttapet. Han hadde en kombinasjon av patologiske leverprøver, koagulopati og anemi. Utslett og gastrointestinale symptomer kunne gi mistanke om cøliaki. Karsinoid syndrom og vaskulitt er tilstander som kan gi et liknende klinisk bilde med kombinasjon av episodiske symptomer fra gastrointestinalkanalen, forhøyet senkning og allmennsymptomer.
På bakgrunn av høy senkning, anemi, vekttap og påvirket allmenntilstand ble pasienten henvist til CT av hals, thorax, abdomen og bekken med intravenøs kontrast. Denne viste små mengder fri væske i bekkenet, splenomegali på 17 cm (lengste diameter), periportalt ødem og markerte lymfeknuter ved leverhilus, hvor den største var 11 mm i kortaksediameter (figur 1).
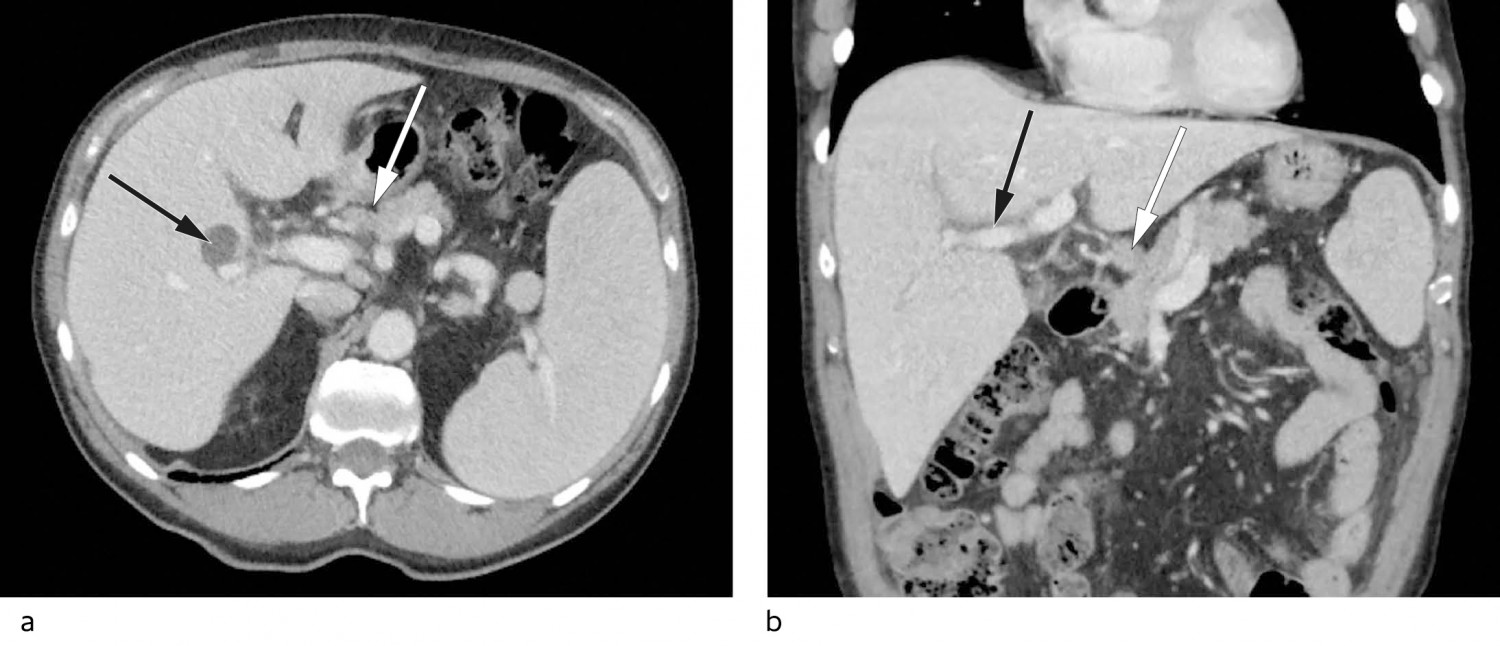
Figur 1 a) CT-bilde i aksialplan av øvre abdomen med forstørret milt og bimilt, markerte lymfeglandler i leverhilus (hvit pil) og en simpel levercyste i høyre leverlapp (sort pil). b) CT-bilde i koronalplan med periportalt ødem (sort pil) og markert lymfeglandel i leverhilus (hvit pil).
Periportalt ødem, som er væskeansamling rundt det portale kretsløpet, er et uspesifikt funn som kan gi mistanke om leverpatologi, men kan også ses ved ekstrahepatiske sykdommer (5). Splenomegali kan ses ved flere sykdomstilstander, eksempelvis portal hypertensjon sekundært til leversykdom, lymfom eller annen hematologisk malignitet. Isolert funn av lett markerte lymfeknuter ved leverhilus uten funn av forstørrede lymfeknuter andre steder gjorde lymfomsykdom mindre sannsynlig. Anemi, påvisbar M-komponent samt venstreforskjøvet blodbilde gav mistanke om hematologisk sykdom.
Få dager etter første konsultasjon ved hematologisk poliklinikk ble det utført beinmargsdiagnostikk. Beinmargsutstryket viste en svært cellerik beinmarg, med normal andel megakaryocytter, lette irregulære trekk i erytropoesen og en tydelig venstreforskjøvet og økt granulocyttopoese, men normal andel plasmaceller. Funnet gav mistanke om en myeloproliferativ sykdom. Cristabiopsi viste en maksimalt cellerik beinmarg med multiple, til dels atypiske mastcelleinfiltrat (figur 2). I tillegg var margen generelt hypercellulær med dominerende myelopoese og megakaryocytter med noe avvikende morfologi. Immunhistokjemi viste at mastcellene var positive for mastcelletryptase, CD117 og CD25. Genetisk analyse viste positiv aktiverende mutasjon i KIT-genet (D816 V). I tillegg ble det tatt prøve for tryptase, en mastcellemediator, som viste forhøyet verdi i serum, 116 mcg/l (< 20).
Beinmargsbiopsien bekreftet diagnosen systemisk mastocytose assosiert med annen hematologisk neoplasi. Systemisk mastocytose er en sykdom som kan gi infiltrasjon av mastceller i flere organer, og på bakgrunn av hudutslett og gastrointestinale symptomer ble han ytterligere utredet.
To måneder senere tok pasienten en stansebiopsi av utslettet hos hudlege. Biopsien viste mastcelleinfiltrasjon i dermis. Det ble utført en gastroskopi, som viste normale makroskopiske funn. Biopsier av duodenalslimhinne viste for det meste normale funn, men det var et lite fokus med tettliggende og lett atypiske mastceller som ble bekreftet med immunhistokjemisk farging (figur 2). KIT-D816 V-mutasjon ble også bekreftet i hud og blod.
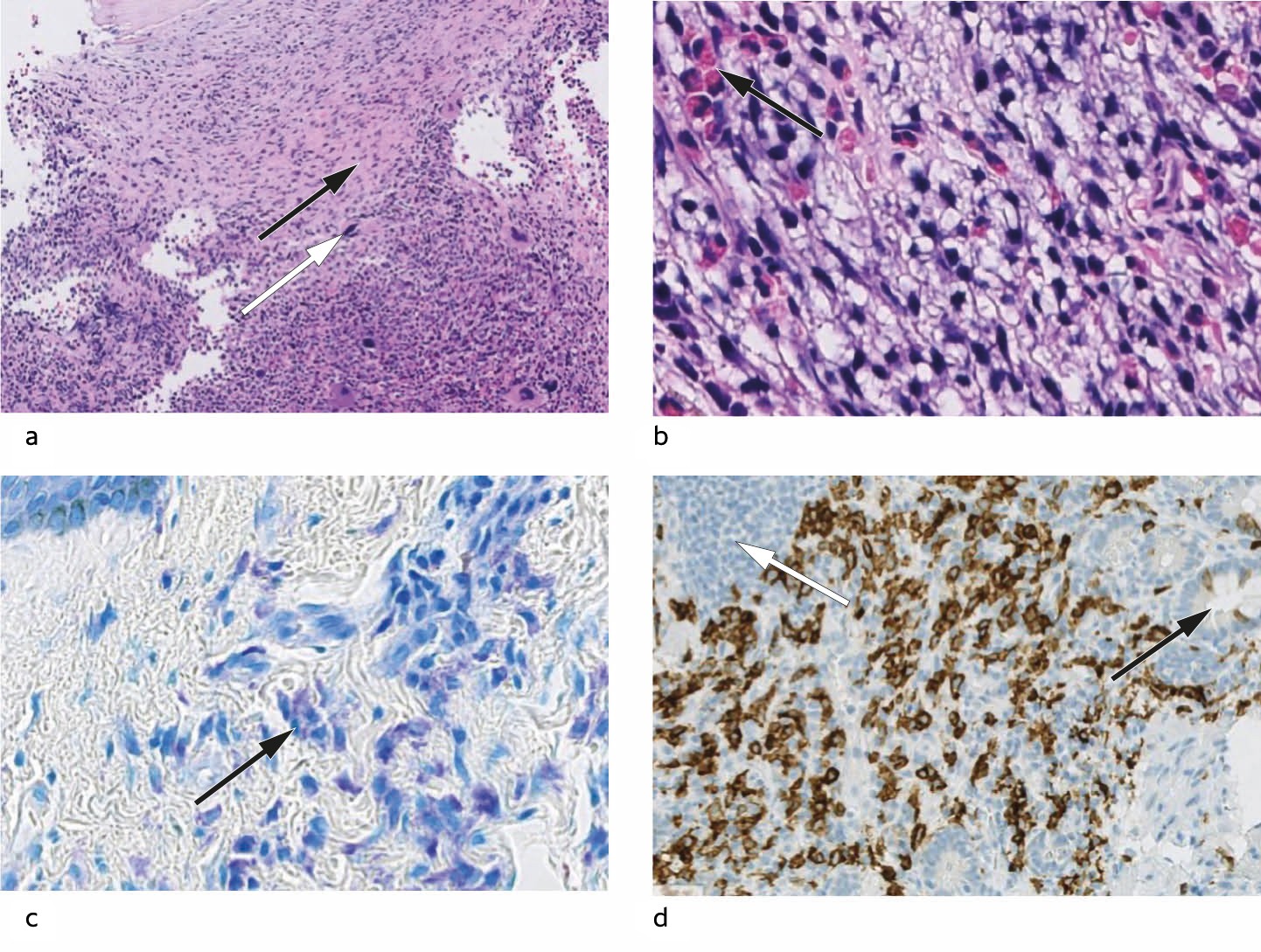
Figur 2 a) Beinmargsbiopsi. Margen er maksimalt cellerik. I øvre halvdel ses mastcelleinfiltrat assosiert med fibrose (svart pil). I nedre halvdel ses hyperplastisk hematopoese, med avvikende megakaryocytter (hvit pil) (HE 150x). b) Beinmargsbiopsi. Neoplastiske mastceller med lyst cytoplasma og hyperkromatiske og irregulære kjerner. Man ser tilblanding av eosinofile granulocytter (sort pil) (HE 400x). c) Hudbiopsi. Giemsafarget snitt viser ansamlinger av mastceller med typiske basofile granula i cytoplasma i øvre del av dermis (sort pil). Basis av epidermis ses øverst i venstre hjørne (400x). d) Duodenalbiopsi. I rutinefarget snitt var det vanskelig å påvise mastcelleinfiltrat, men immunhistokjemisk farging for CD117 viste membranøs brunfarging av tallrike mastceller i basis av slimhinnen. Krypter ses til høyre (sort pil), og en normal lymfefollikkel øverst til venstre (hvit pil).
Pasienten hadde en aggressiv form for systemisk mastocytose. Det var indikasjon for sykdomsmodulerende behandling hovedsakelig på bakgrunn av anemi, som skyldtes mastcelleinfiltrasjon i beinmargen. Han hadde også nylig fått påvist folsyremangel og hadde konstitusjonelle symptomer som sannsynlig skyldtes sykdommen.
Pasienten fikk symptomlindring ved hjelp av histaminantagonist (cetirizin), antihistaminerg syresekresjonshemmer (ranitidin) og protonpumpehemmer (omeprazol). Han fikk også behandling med midostaurin, en kinasehemmer som også hemmer KIT. Initialt ble sykdommen stabilisert, men etter syv måneder ble pasienten dårligere med behandlingskrevende anemi, ascites og vekttap. Han fikk da seks sykluser med kladribin, og responderte med redusert behov for erytrocyttransfusjoner, stabilisering av vekt, mindre uttalt ascites og fall i tryptasenivået.
Pasienten har nå blitt observert i åtte måneder hvor han ikke har fått behandling. Han har ved én anledning blitt innlagt på sykehuset for infeksjon med ukjent fokus. Han tappet da ascites og fikk transfusjonsbehandling med erytrocytter. Utover dette bor han hjemme alene, fyller kriteriene for WHO-funksjonsklasse 1 og har for tiden ingen uttalte plager relatert til sin systemiske mastocytose.
Diskusjon
Systemisk mastocytose ble for første gang beskrevet i Frankrike i 1930-årene (6). Forekomsten er usikker (7). En retrospektiv kohortstudie fra Danmark av voksne diagnostisert med systemisk mastocytose viste en årlig insidens på 0,89 per 100 000 (8). Det utarbeides nå pasientregistre for å få korrekte data på insidens og prevalens (9).
Pasienter med sammensatte sykdomsbilder, som vår pasient, vil ofte kreve en tverrfaglig tilnærming for å komme fram til riktig diagnose. Symptomer og funn ved mastocytose skyldes en kombinasjon av mastcelleaktivering med frigjøring av mastcellemediatorer og organinfiltrasjon. Diagnosen bekreftes ved biopsi og genetisk analyse med henblikk på KIT-mutasjon. Tryptase, et enzym lokalisert til mastcellegranula, kan gi diagnostisk informasjon og kan brukes i oppfølging, da økende nivå er assosiert med større mastcellebyrde og sykdomsprogrediering (10).
Mastocytose blir delt inn i tre grupper: mastcellesarkom, kutan mastocytose eller systemisk mastocytose (9). Mastcellesarkom er en svært sjelden og malign form for solid svulst med evne til destruktiv infiltrasjon og metastasering. Ved kutan mastocytose er de patologiske mastcellene isolert i huden og gir varianter av utslett, hvor urticaria pigmentosa, et makulopapuløst utslett, er det vanligste (9). Når sykdommen kan identifiseres i et organ utover hud, defineres den som systemisk hvis kriteriene i figur 3 er oppfylt (11). Vår pasient oppfylte hovedkriteriet og tre underkriterier. Beinmargen er det vanligste predileksjonsstedet (6), men mastcelleinfiltrasjon kan også forekomme i leveren, milten, gastrointestinalkanalen, skjelettet samt andre organsystemer. Forekomsten i de ulike organene er ukjent (9). Pasienten vår hadde ikke tegn til skjelettaffeksjon på CT-bildene. Hans initiale ryggsmerte var sannsynligvis uten relasjon til mastocytosen.
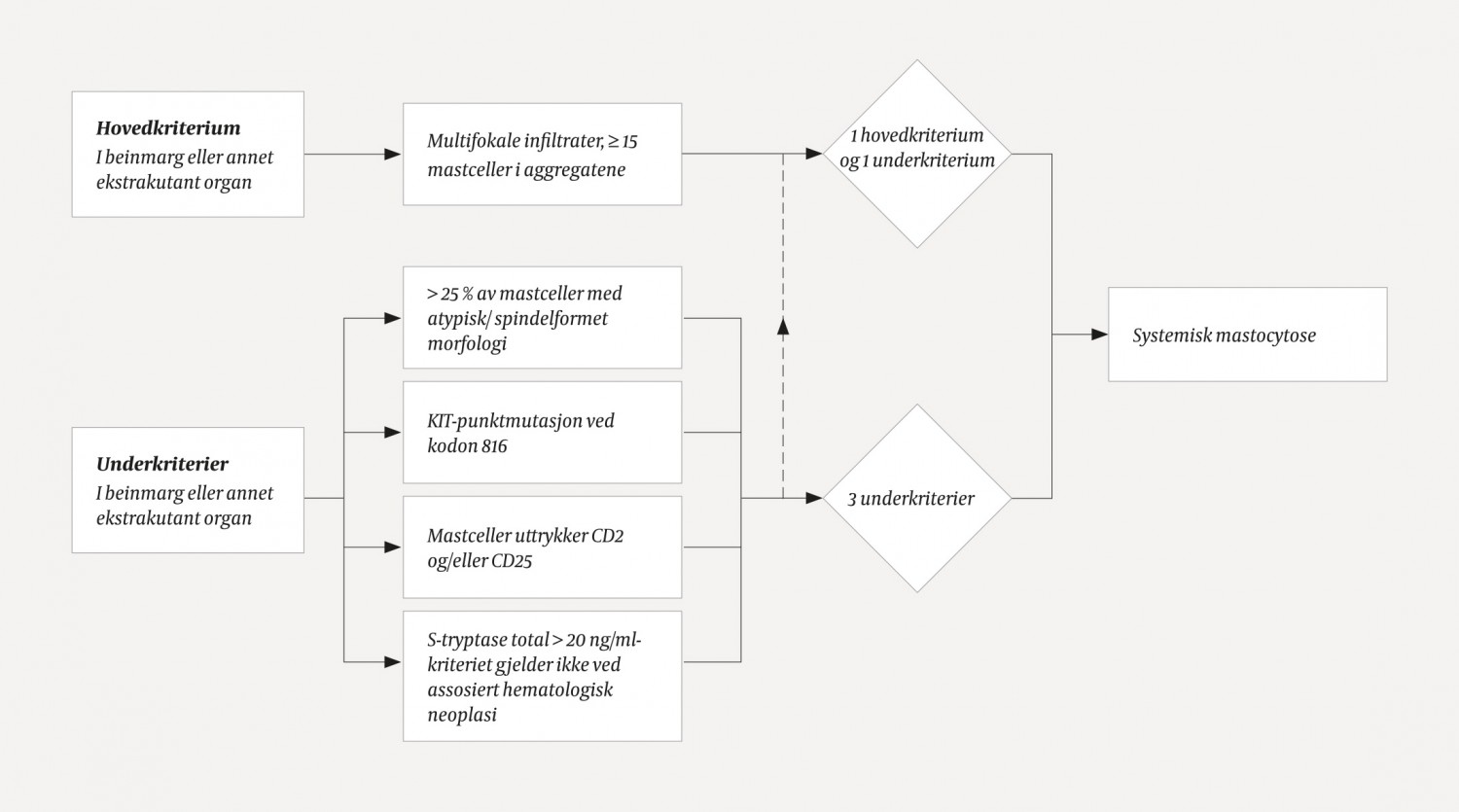
Figur 3 Diagnostisk algoritme for systemisk mastocytose. Diagnosen krever ett hovedkriterium pluss ≥ 1 underkriterium eller ≥ 3 underkriterier. Kriteriene er definert av Verdens helseorganisasjon i 2001 og sist oppdatert i 2016 (11).
Mastocytose tenkes å ha sitt opphav fra muterte hematopoetiske stamceller (12). Studier har vist at > 90 % av pasientene har en aktiverende mutasjon i genet KIT, der over 80 % av disse vil ha en bestemt punktmutasjon i ekson 17, KITD816 V (13, 14). KIT er en transmembran tyrosinkinasereseptor for stamcellefaktor CD117 (6, 13). Mutert KIT leder til en liganduavhengig konstitutiv aktivering og autofosforylering av tyrosinkinase (15) og stimulerer mastcellene til proliferasjon og overlevelse (16).
Tidligere ble systemisk mastocytose klassifisert som en variant av myeloproliferativ neoplasi (17). I henhold til WHO-klassifikasjonen fra 2016 regnes nå tilstanden som en egen sykdomsentitet på grunn av unike kliniske og patologiske trekk med et svært variabelt sykdomsforløp (18). Indolent systemisk mastocytose har god prognose med nærmest normal forventet levetid (9). Kliniske funn som er relatert til organskade forårsaket av mastcelleinfiltrasjon blir definert som C-funn (9, 19). Systemisk mastocytose defineres som aggressiv ved tilstedeværelse av minst ett C-funn og har en dårligere prognose (10).
Hovedfokus for behandling av indolent systemisk mastocytose er symptomhåndtering og monitorering (6). Denne baserer seg i all hovedsak på bruk av antihistaminer, antihistaminerge syresekresjonshemmere, protonpumpehemmere, leukotrienantagonister, kromoglikat og eventuelt steroider (6, 9, 20, 21). Pasienter med systemisk mastocytose har også risiko for episoder med uttalt mastcelleaktivering og utslipp av større mengder histamin og andre inflammasjonsmarkører. Triggere kan være emosjonelt stress, traumer, infeksjoner, allergiske reaksjoner og medikamenter som for eksempel acetylsalisylsyre, ikke-steroide antiinflammatoriske midler (NSAID), opiater, antikolinergika og enkelte kontrastmidler (6, 21).
Vår pasient ble vurdert å ha aggressiv mastocytose, selv om det var vanskelig å si i hvilken grad hans assosierte hematologiske neoplasi, i form av ikke-klassifiserbar myeloproliferativ sykdom, bidro til sykdomsbildet. En godt akseptert standard er at systemisk mastocytose og annen assosiert hematologisk sykdom skal behandles uavhengig av hverandre (9). Behandling av aggressiv systemisk mastocytose med sykdomsmodulerende behandling forblir en utfordring. Flere pasienter responderer på kladribin eller interferon, men de fleste får tilbakefall eller har resistent sykdom (20). I en studie med totalt 22 pasienter viste kladribin en responsrate på 50–55 % hos pasienter med avansert mastocytose. Svært god (major) respons, definert som fullstendig fravær av minst én tidligere mastocytoserelatert organskade, ble oppnådd hos 37 % (22).
Tyrosinkinasehemmeren midostaurin er vist å være effektiv til å hemme aktivert kinase KIT (23). Men selv om initial sykdomskontroll oppnås, ser man ikke en vedvarende remisjon hos alle pasientene. En studie med midostaurin som inkluderte 116 pasienter hvor alle hadde avansert systemisk mastocytose, viste en total responsrate på 60 %, med svært god respons hos 45 % av pasientene (23). Medikamentet ble ikke testet opp mot annen behandling, og effekten var forbigående hos majoriteten av pasientene, med median progresjonsfri overlevelse på 14,1 måneder (23). Andre medikamenter er under utprøving i ulike fasestudier, bl.a. alternative KIT-inhibitorer, med tanke på å oppnå sykdomskontroll (6). Hos unge pasienter med aggressiv sykdom bør en vurdere stamcelletransplantasjon, da dette er eneste mulighet for vedvarende respons og kurasjon (6, 9). Genetiske analyser med neste generasjons sekvensering vil kunne bidra til bedre prognostisk evaluering og mer skreddersydd behandling til pasienter med systemisk mastocytose (6, 24, 25).
Systemisk mastocytose kan affisere mange organsystemer. Spesielt ved diffuse symptomer kombinert med hudutslett er systemisk mastocytose en differensialdiagnose. Sykdommen bør mistenkes ved uvanlig, repetert eller anfallsvis anafylaktoid reaksjon, rødme og synkope. Hos slike pasienter bør huden inspiseres for det karakteristiske utslettet urticaria pigmentosa (kutan mastcelleinfiltrasjon). Forhøyet tryptase og eventuell eosinofili styrker mistanken om systemisk mastocytose (26). På grunn av risiko for anafylaktoide reaksjoner hos pasienter med systemisk mastocytose anbefales adrenalinpenn til alle, og helsepersonell må være klar over risikoen hos denne pasientgruppen.
