Atferdsendring og vrangforestillinger i unge år er oftest et sikkert tegn på schizoid personlighet. Langvarig behandling med antipsykotiske legemidler kan føre til alvorlige dyskinesier. Psykiatrisk sykdom utelukker imidlertid ikke somatisk årsak. Atypisk forløp av psykisk sykdom bør lede oppmerksomheten mot andre differensialdiagnoser.
En kvinne midt i 20-årene utviklet paranoide vrangforestillinger og ble innlagt i psykiatrisk sykehus. Hun hadde vært frisk utenom det aktuelle, og det var ingen kjente arvelige lidelser i familien. Hun hadde friske foreldre, som hadde felles slektninger noen generasjoner tilbake, og to eldre, friske søsken. Svangerskap, fødsel og barne- og ungdomstid hadde vært uten anmerkning. Hun gjennomførte videregående skole med middels gode prestasjoner og gikk deretter ut i yrkeslivet.
Vedvarende paranoide vrangforestillinger og personlighetsendringer førte til at hun ganske snart fikk diagnosen schizofreni. Det ble startet medisinering med ulike nevroleptika (zuklopentixol, olanzapin, klozapin), uten tilfredsstillende effekt på symptomene. Allerede 1 – 2 år etter oppstart med zuklopentixol ble det bemerket orofaciale dyskinesier, oppfattet som tardive dyskinesier.
De tardive dyskinesiene ble antatt å være bivirkning av de antipsykotiske legemidlene hun brukte. Slike symptomer bør medføre seponering eller skifte til et annet nevroleptikum med mindre antidopaminerg effekt. Symptomene kan persistere etter seponering.
Medisineringen ble lagt om til olanzapin, som senere ble seponert pga. manglende effekt på de psykiske symptomene. Overgang til klozapin førte heller ikke til endring av de orofaciale dyskinesiene. Etter hvert ble det også involuntære bevegelser i ekstremitetene, og gangmønsteret endret seg. De nevrologiske symptomene ble mer fremtredende.
Tardive dyskinesier er vanligvis begrenset til gjentatt og påfallende ansiktsgrimasering. I dette tilfellet økte de ufrivillige bevegelsene over tid, til tross for medikamentomlegging. Etter hvert omfattet de også ekstremitetene. Dette endret pasientens gangmønster. Det kliniske bildet var nå lite forenlig med schizofreni. Hun ble derfor søkt innlagt i nevrologisk avdeling til videre utredning.
Ved klinisk undersøkelse gjorde hun greit rede for sin situasjon, men viste klar svikt av kognitive funksjoner, med ukritisk språkbruk og atferd. Det forelå uttalt dysartri og nærmest kontinuerlige orofaciale dyskinesier med grimasering og tungeprotrusjon. Pasienten hadde skrevet små brev og dikt til sin behandler, der hun skrev om tungen som skjøv «tyggisen» ut av munnen. Hun beskrev også et sterkt ønske om å kunne spise normalt, slik at hun kunne sitte til bords sammen med andre. Det var bittmerker i tungen, og pasienten hadde betydelige svelgevansker. Muskulaturen var hypoton, og man observerte koreiforme involuntære bevegelser i alle ekstremiteter. Sensibiliteten var intakt, så langt den lot seg undersøke. De dype senerefleksene var middels livlige, bortsett fra akillessenerefleksene, som var utslukket. Gangen var ataktisk og preget av koreiforme bevegelser i truncus og dels i ekstremitetene – hun hadde en slentrende og veivende gangart. Det var upåfallende organstatus for øvrig, bortsett fra obstipasjon og oppblåst tarmsystem.
De økende nevrologiske symptomene, med svelgevansker, tungebiting, dysartri og hypoton muskulatur, i tillegg til betydelige bevegelsesforstyrrelser, var lite forenlig med tardive dyskinesier. Diagnosen Huntingtons sykdom, som medfører både chorea, dystonier og kognitiv svikt, ble vurdert, men hun hadde ingen familiehistorie med denne arvelige sykdommen.
Rutineblodprøvene var normale, inkludert ceruloplasmin og lipidelektroforese. Urinen var fysiologisk. Gentest for Huntingtons sykdom var negativ. Det forelå signifikant økning av kreatinkinase (CK) til 495 IE (normalt < 150 IE) i blod. EEG var lett patologisk, med økt mengde langsom aktivitet.
Det kliniske bildet med debut av nevropsykiatriske symptomer i tredje dekade hos en tidligere frisk kvinne, koreatiske bevegelser, grimasering, ataktisk gange og tydelige tegn til lett polynevropati ga mistanke om koreoakantocytose, en hereditær, progredierende nevrodegenerativ tilstand som affiserer flere organsystemer. Det kliniske bildet og karakteristiske funn ved blodutstryk er avgjørende for diagnosen.
I perifert blodutstryk var det et signifikant økt antall akantocytter. I preparert utstryk etter fire timer hadde pasienten 3,2 % akantocytter, mens det i prøver fra to friske kontrollpersoner var 0 %. Cerebral MR viste lett bilateral atrofi av caput nucleus caudatus (fig 1) og i tillegg en lett grad av sentral og kortikal atrofi, alderen tatt i betraktning. Western blot, utført langt senere, viste totalt fravær av proteinet chorein.
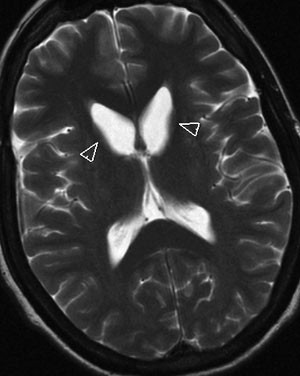
Figur 1 Aksialt T2-vektet bilde som viser atrofi av caput nucleus caudatus bilateralt (pilhode)
Funnene bekreftet diagnosen koreoakantocytose. Dette er en av flere tilstander som faller inn under betegnelsen nevroakantocytose. Diagnosen baseres på de nevrologiske utfall kombinert med påvisning av akantocytter i perifert blod.
Diskusjon
Vår pasient anskueliggjør det diagnostiske dilemmaet som oppstår når en sjelden nevrodegenerativ lidelse starter med vanlig forekommende psykiatriske symptomer. Den somatiske undersøkelse viste at det i dette tilfellet forelå såkalt koreoakantocytose (OMIM 200150). Koreoakantocytose faller inn under begrepet nevroakantocytose, som bl.a. også innbefatter abetalipoproteinemi (OMIM 200100), hypobetalipoproteinemi (OMIN 107730) og McLeods syndrom (OMIN 314850).
Akantocytter er røde blodceller med uregelmessige, butte utløpere med varierende bredde og lengde (acantha = torner). Utløperne i akantocyttene skyldes en defekt i cellemembranen (fig 2). De ble første gang beskrevet i 1950 ved tilstanden abetalipoproteinemi (Bassen-Kornzweigs syndrom) (1). Begrepet «nevroakantocytose» ble første gang brukt i 1967 om en tilstand med nevrologiske symptomer og akantocytter i perifert blod, uten abetalipoproteinemi (2). Akantocyttene skilles fra echinocyttene («piggepler») ved at de sistnevnte har beholdt sin runde form og utløperne («piggene») er jevnt fordelt ut over hele overflaten. Andelen akantocytter i blodet varierer betydelig hos pasienter med nevroakantocytose, uten at mengden ser ut til å korrelere med sykdommens alvorlighetsgrad (2). Det er også beskrevet tilfeller med et klinisk bilde av nevroakantocytose uten at akantocytter er påvist hos pasienten. Påvisning av akantocytter er diagnostisk viktig ved denne tilstanden og krever spesialfarging og hematologisk kompetanse. Det er ikke tilstrekkelig med ett enkelt perifert blodutstryk. Prepareringen er tidkrevende og går over mange timer.
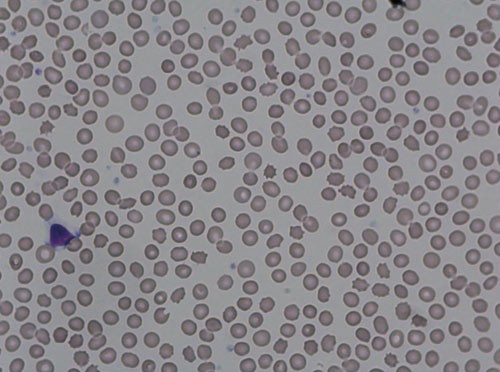
Figur 2 Akantocytter i perifert blod fra pasienten, karakteristisk for chorioakantocytose
Begrepet «nevroakantocytose» omfatter en heterogen gruppe tilstander. Det er to relativt klare undergrupper, nemlig nevroakantocytose med og nevroakantocytose uten forstyrrelser i lipoproteinstoffskiftet. I tillegg til disse er det beskrevet en rekke sykdomsbilder der man har funnet akantocytter i perifert blod, kombinert med nevrologiske funn (3). Undergruppen der man finner forstyrrelser i lipoproteinstoffskiftet og akantocytter i perifert blod er assosiert med malabsorpsjon av lipider. Nevrologisk ser man affeksjon av medulla, retina og det perifere nervesystemet. I denne gruppen finner vi tilstandene abetalipoproteinemi, familiær hypobetalipoproteinemi, Andersons sykdom og atypisk Wolmans sykdom.
Nevroakantocytose uten forstyrrelser i lipoproteinstoffskiftet affiserer primært hjernen, spesielt basalgangliene, og er assosiert med nevropsykiatriske symptomer, progredierende kognitiv svikt og koreatiske bevegelser. Tilstanden kan minne om Huntingtons sykdom.
Koreoakantocytose er en form for nevroakantocytose som vanligvis arves autosomalt ressesivt. Chorea er ufrivillige, kastendebevegelser som kan forekomme både proksimalt og distalt i ekstremitetene. Tilstanden kjennetegnes av bevegelsesforstyrrelser, vanligvis hyperkinetiske, i kombinasjon med nevropsykiatriske symptomer og akantocytter i perifert blod. Dette er en primært nevrodegenerativ tilstand som affiserer flere organsystemer. Det er beskrevet ulike debutsymptomer, hvorav chorea, epilepsi og nevropsykiatriske symptomer er de vanligste. Det kan gå flere år før affeksjon av flere organsystemer avdekkes (4), noe som gjør diagnostikken vanskelig. Sykdommen debuterer typisk i 20 – 40 års alder, men både tidligere og senere debut er beskrevet (3, 5).
Det er påvist en rekke mutasjoner i VPS13A-genet på kromosom 9q21 (6, 7). Genet uttrykkes i alle affiserte vev. VPS13A koder for to proteiner med henholdsvis 3 095 og 3 174 aminosyrer. Det lengste proteinet har fått betegnelsen chorein (8). Mangel på dette proteinet er sannsynligvis primær sykdomsårsak. Bestemmelse av chorein gjøres kun når den kliniske diagnose er rimelig sikker. Man vet lite om den normale funksjonen til dette proteinet.
Bevegelsesforstyrrelser affiserer de fleste av pasientene med koreoakantocytose. Ufrivillige bevegelser i tunge, ansiktet, pharynx og larynx er karakteristisk. Selvmutilerende atferd med stadig biting i leppe og tunge ses regelmessig (9). Bruksisme, dvs. gnissing og pressing av tennene, ufrivillig raping og spytting forekommer. Vokalisering er til stede hos to tredeler av pasientene. Dysfagi er et resultat av dystone tungebevegelser og kan føre til mangelfullt næringsinntak. Progredierende dysartri er vanlig og kan være debutsymptom. En del pasienter utvikler mutisme.
De ufrivillige bevegelsene er vanligvis koreatiske, men pasienten kan også utvikle parkinsonisme og dystoni (5). Chorea og dystone bevegelser gir et tydelig avvikende gangmønster. Pasienten går ustødig, med lange skritt og med veivende armbevegelser. På den måten skiller de seg klart fra det man vanligvis ser ved Huntingtons sykdom. Epilepsi, oftest med generaliserte anfall, ses i forløpet hos nær halvparten av de affiserte pasientene, og kan også forekomme som debutsymptom (9). Nevrologisk undersøkelse avdekker myopati, som gir pareser og atrofi hos omtrent halvparten av pasientene. Forhøyet serumkonsentrasjon av kreatinkinase ses hos et flertall av pasientene og kan være til stede før de nevrologiske symptomene blir åpenbare. Perifer nevropati er et obligat tegn. Sensoriske utfall kan ses, ofte begrenset til tap av vibrasjonssans. Unormale øyebevegelser er beskrevet som en del av sykdomsbildet, noe som sannsynliggjør at nevrodegenerasjonen ved koreoakantocytose også omfatter hjernestammen.
Det er personlighetsendringer – pasienten blir kognitivt svekket og kan ha psykiatriske symptomer i form av trikotillomani, tvangspreget handlingsmønster, AD/HD-liknende symptomer og symptomer som kan minne om Tourettes syndrom. Personlighetsendring kan være debutsymptom. Det kliniske bildet er altså meget heterogent, og det forekommer betydelig fenotypisk variasjon selv innenfor samme familie (9). Bildediagnostisk finner man ofte lett atrofi av nucleus caudatus og av putamen, som ses best i koronalsnitt (10).
Vår pasienthistorie minner oss om at psykiatrisk sykdom ikke utelukker somatiske årsaksforhold. Den aktuelle pasienten debuterte med et bilde som primært pekte mot en psykiatrisk tilstand, og man tolket de ufrivillige bevegelsene som tardive dyskinesier. Alle pasientens symptomer, som til sammen er karakteristiske for koreoakantocytose, var beskrevet i den psykiatriske journalen, men koblingen til en så sjelden sykdom er avhengig av kunnskap om den. Psykiaterne hadde søkt etter årsaken til hennes sykdomsbilde både i psykopatologiske og farmakologiske forhold, men henviste videre til nevrologisk utredning for å søke etter andre forklaringer.
Koreoakantocytose er en uhelbredelig og progredierende tilstand. For vår pasient har dette ført til heldøgns omsorgsbehov. Det er viktig for både pasient, pårørende og helsepersonell at korrekt diagnose stilles så tidlig som mulig. Psykiatriske diagnoser fører hyppig til at både pårørende og helsepersonell leter i pasientens bakgrunn etter utløsende faktorer. Selvbebreidelse hos pårørende er ikke uvanlig. Når en arvelig tilstand avdekkes, fritas både pårørende og helsepersonell fra å lete etter psykososiale årsaksforhold, og veiledning og problemløsning blir mer realistisk. Videre kan omsorgstilbudet tilpasses pasientens behov og prognosen når man er kjent med sykdommens forløp. Den medisinske behandlingen er symptomatisk. Erfaringsmessig er de nevropsykiatriske symptomene mer tilgjengelige for behandling enn de involuntære bevegelsene (11).
Koreoakantocytose er en ekstremt sjelden tilstand, men er tidligere beskrevet fra Norge (9). Sykdommens ulikeartede debutsymptomer fører ofte til at den kan være vanskelig å diagnostisere. Vår pasient debuterte med et bilde av en schizofreniform psykose og ufrivillige bevegelser som kunne være forenlig med tardive dyskinesier. Personlighetsendring og ufrivillige bevegelser som ikke var typiske for bruk av nevroleptika førte til at man måtte søke andre organiske årsaker til pasientens sykdomsbilde.
