Immunglobulin G subklasse 4 (IgG4)-relatert sykdom er en relativt nyoppdaget immunmediert systemsykdom som kjennetegnes av inflammasjon og progredierende fibrose. Pankreatitt og sialadenitt er de vanligste kliniske manifestasjonene, men sykdommen kan ramme nesten alle kroppens organer. Ved mikroskopering av biopsi ses inflammasjon med en betydelig andel IgG4-positive plasmaceller og et karakteristisk fibrosemønster. Glukokortikoider er førstevalg i behandling.
IgG4-relatert sykdom er en sjelden og relativt nyoppdaget tilstand, som siden 2003 er blitt ansett som en immunmediert systemsykdom. Sykdommen kan ramme tilnærmet alle kroppens organer og strukturer og kan prinsipielt sammenlignes med sarkoidose: en inflammatorisk tilstand med et heterogent symptombilde som kjennetegnes av karakteristiske histopatologiske funn i affiserte organer.
Ved mikroskopering av vevsbiopsi ses lymfoplasmacytære infiltrater og et karakteristisk sirkulært fibrosemønster, såkalt storiform fibrose (fig 1a). En stor andel av de infiltrerende plasmacellene uttrykker B-cellereseptorer av IgG4-subklassen (såkalte IgG4-positive plasmaceller), hvilket har gitt sykdommen dens navn. Massiv infiltrasjon av immunceller i parenkym og vener kan føre til henholdsvis tumordanning og oblitererende flebitt i affiserte organer (1).
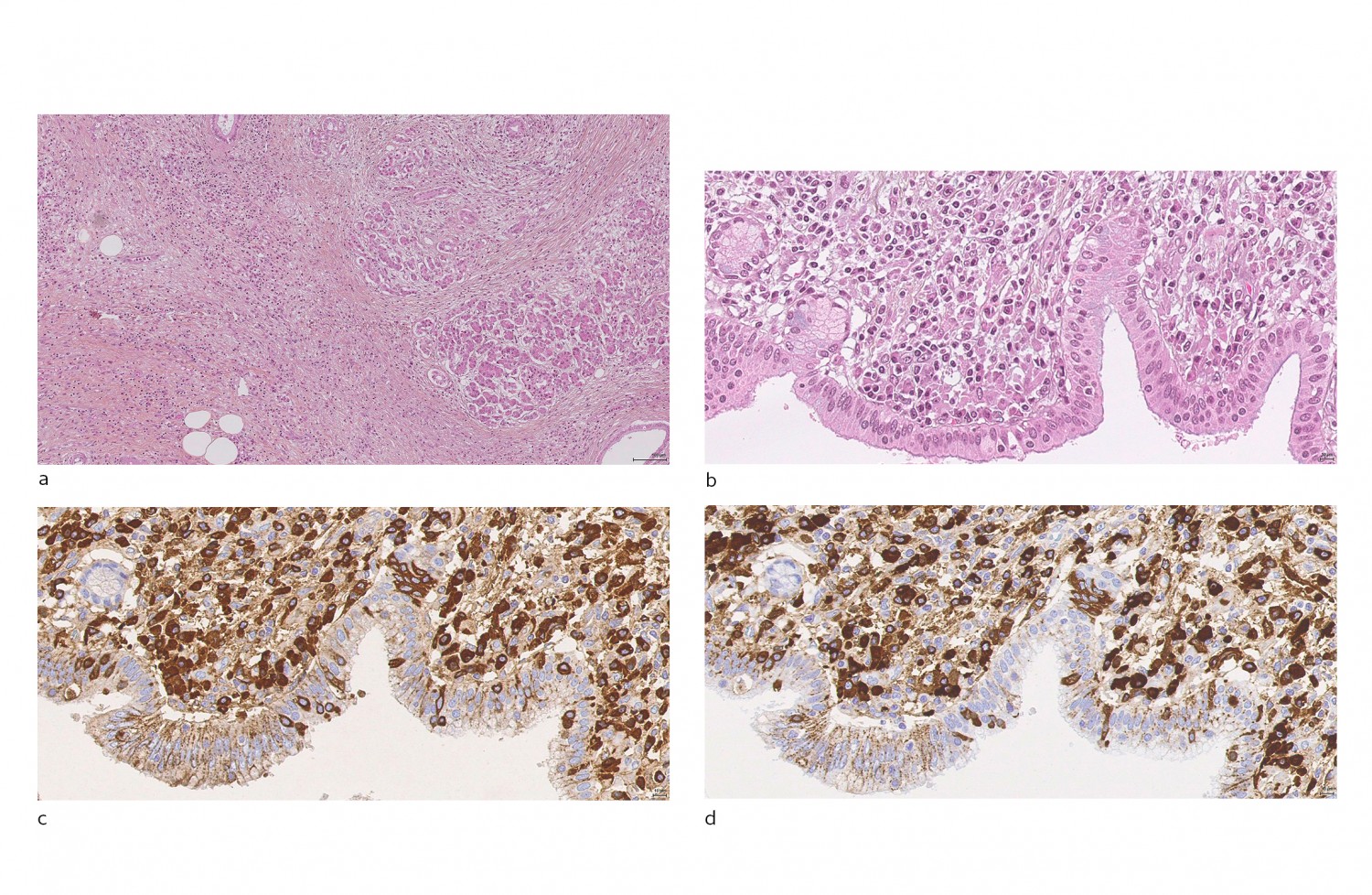
Figur 1 Histologi fra pasient med IgG4-relatert sykdom med pancreasaffeksjon. a) Snitt fra pancreas. Hematoksylin- og eosinfarge (HE). 20x forstørrelse. Storiform fibrose og lymfoplasmacytær betennelse omkring rester av bevart acinært pancreasvev, fett og enkelte utførselsganger. b) Pancreas. HE. 80x forstørrelse. Utførselsgang med uttalt subepitelial lymfoplasmacytær betennelse.c) Pancreas. Immunhistokjemisk farging for IgG. 80x forstørrelse. Flertallet av de subepiteliale plasmacellene er IgG-positive (brun farge). d) Pancreas. Samme snitt som (c). Immunhistokjemisk farging for IgG4. 80x forstørrelse. Nærmest alle de IgG-positive plasmacellene er også positive for IgG4 (brun farge)
Epidemiologien er kun delvis kartlagt, men prevalensen er angitt til 0,28 – 1,08 tilfeller per 100 000 (2). Gjennomsnittsalderen ved diagnosetidspunktet er 60 år. Menn rammes hyppigere enn kvinner, særlig ved pankreatikobiliær sykdom, hvor kjønnsforholdet er 3:1. Hos pasienter med affeksjon av spyttkjertler er kjønnsforskjellene mindre uttalt (1). Årsaken til kjønnsmessige forskjeller i organaffeksjon er foreløpig ukjent. Tilstanden er beskrevet i en kasuistikk i Tidsskriftet (3).
Bakgrunn
I 2001 fant Hamano og medarbeidere forhøyet serum IgG4-nivå hos 20 pasienter med autoimmun pankreatitt sammenlignet med kontrollgruppen (4). To år senere ble det i samme pasientgruppe vist lymfoplasmacytær betennelse med høye nivåer av IgG4-positive plasmaceller i pankreasbiopsier samt ekstrapankreatiske lesjoner med tilsvarende histopatologisk og immunhistokjemisk fenotype (5). Dette ledet til hypotesen om en bakenforliggende systemsykdom med multiorganaffeksjon. I ettertid har man observert slike funn i tilnærmet alle organer og strukturer, inkludert galleveier, aorta, meninger, spyttkjertler, orbita, lunger, nyrer, retroperitoneum og lymfeknuter.
I 2011 ble begrepet IgG4-relatert sykdom introdusert (6), og omfatter nå en rekke tilstander som tidligere har vært ansett som enkeltstående, idiopatiske entiteter, inkludert autoimmun pankreatitt, Mikulicz’ sykdom (skleroserende sialodakryoadenitt), Riedels tyreoiditt og Ormonds sykdom (idiopatisk retroperitoneal fibrose) (ramme 1). Disse tilstandene representerer forskjellige manifestasjoner av den samme fibroinflammatoriske systemsykdommen; IgG4-relatert sykdom.
Ramme 1 Et utvalg av tilstander som nå regnes som del av IgG4-relatert sykdom. Omarbeidet etter Kamisawa og medarbeidere 2015 (1)
Immunglobuliner
Plasmaceller utgår fra aktiverte B-lymfocytter og produserer og utskiller immunglobuliner (antistoffer). En immunglobulinmonomer består av to tunge og to lette kjeder som sammen danner to Fab-regioner (Fragment antigen binding) og én Fc-region (Fragment crystallizable) (fig 2).

Figur 2 En immunglobulinmonomer består av to tunge og to lette kjeder som sammen danner to Fab-regioner og én Fc-region
Fab-regionene binder antigen og bestemmer immunglobulinets affinitet og spesifisitet. Alle immunglobuliner som produseres av én enkelt plasmacelle, har identiske Fab-regioner og gjenkjenner samme antigen.
Fc-regionen dikterer immunglobulinets klasse og funksjon. Det finnes fem hovedklasser av immunglobuliner: IgM, IgD, IgG, IgA og IgE. Fc-regionen aktiverer immunforsvarets effektorsystemer, slik som makrofager, nøytrofile celler og komplement. De fem klassene har strukturelt forskjellig Fc-region med varierende evne til å aktivere diverse effektorsystemer og produserer ulik immunrespons.
Hvilken klasse som er optimal, avhenger av hvilken type antigenstimulus kroppen utsettes for. For eksempel vil IgG effektivt bekjempe ekstracellulære bakterier, mens IgE bidrar til et potent forsvar mot parasitter. Omkringliggende aktiverte immunceller, som T-lymfocytter, produserer signalsubstanser (cytokiner) som via parakrin signalisering stimulerer plasmaceller til å skifte Fc-region (fra for eksempel IgM til IgG) til den klassen som er best egnet til å bekjempe det aktuelle patogenet. Siden denne endringen kun skjer i Fc-regionen, vil et slikt klasseskifte ikke påvirke Fab-regionene. Immunglobulinet gjenkjenner stadig det samme antigenet, men resultatet (effekten) av antigenbinding vil optimaliseres slik at de mest fordelaktige effektorsystemene blir aktivert.
Immunglobulin G4
Immunglobulin G har fire subklasser (IgG1, IgG2, IgG3 og IgG4). Disse subklassene har i hovedsak lik Fc-region, men små forskjeller i primærstrukturen gir subklassene ulik funksjon. IgG1 er den dominerende subklassen i serum, mens IgG4 kun utgjør om lag 5 % av den totale IgG-konsentrasjonen hos friske individer (7). Forhøyede serumnivåer av IgG4 ses hos om lag 60 % av pasientene med IgG4-relatert sykdom. Dette er et uspesifikt funn, da IgG4-forhøyelse i serum også finnes ved andre tilstander (7, 8).
IgG1 er et proinflammatorisk immunglobulin som fører til potent aktivering av makrofager og komplement. Dette gjelder også, i varierende grad, IgG2 og IgG3. IgG4 har ikke samme evne til å aktivere effektorsystemer og har derfor begrenset proinflammatorisk effekt. IgG4 kan imidlertid binde seg til, og nøytralisere, Fc-regionen til andre IgG-subklasser (7). IgG4 har derfor en netto antiinflammatorisk effekt, noe som konseptuelt kan virke paradoksalt siden IgG4-relatert sykdom kjennetegnes av nettopp inflammasjon. Dette har utfordret teorien om rollen til IgG4 i sykdommens patogenese.
En teori antyder at de høye nivåene av IgG4-positive plasmaceller er en konsekvens av, og ikke en årsak til, inflammasjonen i IgG4-relatert sykdom (1). Ifølge denne teorien skyldes vevsdestruksjonen en vedvarende aktivering av CD4-positive T-lymfocytter som igjen aktiverer fibroblaster og makrofager med resulterende fibrosedanning. Videre postuleres at utslipp av cytokiner fra aktiverte T-lymfocytter stimulerer lokale plasmaceller til å gjennomgå klasseskifte til IgG4. Ifølge denne hypotesen er IgG4-predominansen derfor sekundært til den underliggende patologiske prosessen. IgG4-relatert sykdom er en hyperinflammatorisk tilstand, og det virker plausibelt at IgG4 ikke er direkte årsaksgivende basert på proteinets manglende proinflammatoriske evner. Denne teorien er blitt utfordret av at det monoklonale anti-CD20-antistoffet rituksimab virker å være en effektiv behandling av IgG4-relatert sykdom. Gjennom antistoff-, celle- og komplementavhengige mekanismer fører rituksimab til selektiv deplesjon av B-lymfocytter, da dette er de eneste cellene som uttrykker CD20-proteinet på cellemembranen. Dette fører til reduserte nivåer av plasmaceller og følgelig nedsatt produksjon av immunglobuliner, i hovedsak IgG og IgM. Hvorfor har da B-lymfocyttdeplesjon terapeutisk effekt dersom sykdommen er mediert av T-lymfocytter, og B-lymfocytter og IgG4 ikke er involvert i patogenesen? En mulig forklaring er at det er antigenpresenterende B-lymfocytter som underholder den vedvarende aktiveringen av T-lymfocyttene, og at deplesjon av slike B-lymfocytter fører til opphør av den patologiske T-lymfocyttresponsen (9). Dette passer med teorien om at immunglobulinene kun er passive deltagere i patogenesen til IgG4-relatert sykdom.
Andre teorier rundt patogenesen til IgG4-relatert sykdom angir mastceller, basofile granulocytter, eosinofile granulocytter og plasmablaster som toneangivende deltagere. Aktivering av makrofager og komplement er også blitt postulert som en sentral mekanisme (8). Som ved andre kroniske inflammatoriske tilstander er det plausibelt at en miljøfaktor som infeksjon hos genetisk predisponerte individer fører til defekt immuntoleranse med resulterende vedvarende immunrespons (1).
Genetiske faktorer er kun delvis kartlagt, men man har identifisert kandidatgener som kan være assosiert med utvikling av IgG4-relatert sykdom med pancreasaffeksjon, inkludert HLA-allelene DRB1*0405 og DRB1*0401, samt polymorfismer i genene FCRL3 og CTLA4 som er involvert i regulering av henholdsvis B- og T-lymfocytter (7, 10).
Patogenesen til IgG4-relatert sykdom er sannsynligvis multifaktoriell og kompleks og bare delvis kartlagt.
Diagnostikk
IgG4-relatert sykdom diagnostiseres på bakgrunn av kliniske, biokjemiske og histopatologiske funn. Eksklusjon av differensialdiagnoser er essensielt, og et utvalg av disse er anført i ramme 2. I 2012 introduserte en internasjonal multidisiplinær ekspertgruppe diagnostiske kriterier (11), hvor antall IgG4-positive plasmaceller per synsfelt (40x) i kombinasjon med lymfoplasmacytær betennelse, storiform fibrose og/eller oblitererende flebitt i det histologiske preparatet brukes til å vurdere sannsynligheten for IgG4-relatert sykdom. Det er også foreslått flere organspesifikke diagnostiske kriterier.
Ramme 2 Et utvalg av relevante differensialdiagnoser
-
Malignitet (lymfom, sarkom, kolangiokarsinom, cancer pancreatis, m.fl.)
Biokjemiske analyser
Forhøyet serum IgG4-konsentrasjon ses hos majoriteten av pasientene, men dette er verken et sensitivt eller spesifikt funn, og må tolkes med forsiktighet. Forhøyet IgG4/total IgG-ratio i serum er angitt å ha noe høyere spesifisitet (1) Serumnivået av plasmablaster (et cellestadium mellom B-lymfocytter og differensierte plasmaceller) er ofte forhøyet og virker å ha en større diagnostisk nytteverdi enn serum-IgG4, men en slik test er per i dag ikke tilgjengelig for klinisk bruk (7). Klinisk erfaring tilsier at pasientene kan ha vedvarende forhøyet C-reaktivt protein og senkningsreaksjon, men disse kan også være i normalområdet (7, 12).
Histologi og immunhistokjemi
Histopatologisk undersøkelse viser lymfoplasmacytær betennelse, og ved immunhistokjemisk undersøkelse finnes en betydelig andel IgG4-positive plasmaceller (fig 1 a-d). Antall IgG4-positive plasmaceller per synsfelt (40x) som kreves for å vurdere diagnosen, avhenger av organet som undersøkes. En ratio mellom IgG4/IgG-positive plasmaceller større enn 0,4 øker ytterligere sannsynligheten for at det kan dreie seg om IgG4-relatert sykdom (11). Storiform fibrose og oblitererende flebitt, hvor infiltrerende plasmaceller oblitererer venelumen, er andre typiske funn.
Bildediagnostikk
Bildediagnostiske undersøkelser evner ikke å skille IgG4-relatert sykdom fra differensialdiagnoser som malignitet. PET-undersøkelse kan være av verdi for å vurdere sykdomsutbredelse og behandlingsrespons (1).
Kliniske fenotyper
IgG4-relatert sykdom regnes som én enkelt systemsykdom med variabel organaffeksjon. Symptomer oppstår gradvis sekundært til ekspansive og/eller okkluderende prosesser, for eksempel eksoftalmus eller obstruktiv icterus. Uspesifikke symptomer som magesmerter, vekttap og fatigue forekommer (1, 13). I en japansk kohortstudie med 235 pasienter med IgG4-relatert sykdom var pankreatitt den vanligste manifestasjonen (60 %), etterfulgt av sialadenitt, tubulointerstitiell nefritt, dakryoadenitt og periaortitt (13). Over halvparten av pasientene hadde affeksjon av to eller flere organer.
Autoimmun pankreatitt
Det finnes to typer autoimmun pankreatitt, type 1 (lymfoplasmacytær skleroserende pankreatitt) og type 2 (idiopatisk duct-centric pankreatitt), hvorav kun type 1 regnes som en manifestasjon av IgG4-relatert sykdom. Typisk presentasjon er magesmerter, eventuelt obstruktiv icterus sekundært til parenkymal fibrose eller ledsagende IgG4-relatert skleroserende kolangitt.
Mikulicz’ sykdom
Skleroserende sialodakryoadenitt kjennetegnes av smertefri forstørrelse av glandulae lacrimalis, parotis og/eller submandibularis med eller uten siccafenomener. I motsetning til Sjögrens syndrom ses mannlig predominans, fravær av anti-SSA- og anti-SSB-autoantistofffer, samt klinisk respons på kortikosteroider.
Tubulointerstitiell nefritt
Tubulointerstitiell nefritt er den vanligste intrarenale manifestasjonen av IgG4-relatert sykdom, men er ofte asymptomatisk og påvises ved målrettet undersøkelse hos pasienter med ekstrarenal IgG4-relatert sykdom (7). Bildeundersøkelser kan påvise renomegali og/eller lavattenuerende renale lesjoner. Hos symptomatiske pasienter varierer de kliniske funnene fra mikroalbuminuri til akutt nyresvikt. I motsetning til medikamentutløst tubulointerstitiell nefritt har pasienter med IgG4-relatert sykdom som regel ikke feber, CRP-stigning, pyuri eller hvite blodlegemesylindre i urinen (12).
Retroperitoneal fibrose og periaortitt
Retroperitoneale organer som uretere og aorta kan affiseres, noe som fører til henholdsvis hydronefrose og periaortitt. Dette ble tidligere omtalt som Osmonds sykdom (idiopatisk retroperitoneal fibrose). Andre årsaker til retroperitoneal fibrose, som medikamenter, infeksjoner og malignitet må utelukkes (7).
Andre manifestasjoner
Pasientene har ofte lokalisert eller generalisert, smertefri lymfadenopati. Feber ses sjelden. Eksoftalmus kan være et resultat av dakryoadenitt eller orbitale pseudotumores (1). Hypofyseaffeksjon kan gi hormonavvik og ekspansive komplikasjoner som hodepine og bitemporal hemianopsi (7). Affeksjon av cerebrum er uvanlig, men diffus fortykkelse av meningene (hypertrofisk pakymeningitt) er beskrevet og kan gi symptomer i form av hodepine, radikulopati eller hjernenervepareser (7).
Hvem skal utredes?
Indikasjon for utredning for IgG4-relatert sykdom vurderes på individuelt grunnlag, gjerne i samråd med revmatolog. Pasienter med følgende kliniske presentasjoner bør vurderes utredet: pankreatitt uten åpenbar årsak, skleroserende kolangitt, bilateral forstørrelse av spytt- eller tårekjertler, retroperitoneal fibrose, orbitale pseudotumorer og/eller eksoftalmus.
Behandling
Førstevalget i behandling av IgG4-relatert sykdom er høydose peroral prednisolon (0,6 mg/kg) i 2 – 4 uker, etterfulgt av gradvis dosereduksjon til vedlikeholdsdose på 2,5 – 5 mg daglig. Varighet av vedlikeholdsbehandling er ikke definert. De fleste pasientene har en rask klinisk respons på kortikosteroider (7), men residiv i forbindelse med dosereduksjon eller seponering ses hyppig. Kortikosteroider reduserer inflammasjon og kan føre til bedring av vevsfunksjon. Tidlig behandling er avgjørende for å unngå progredierende fibrose med irreversibelt tap av organfunksjon. Det naturlige forløpet hos ubehandlede pasienter er ikke kartlagt.
Prospektive studier på bruk av tradisjonelle steroidsparende medikamenter som metotreksat og azatioprin finnes ikke. Rituksimab er som nevnt blitt brukt, men randomiserte studier kreves for å etablere optimal behandlingsstrategi.
Oppsummering
IgG4-relatert sykdom er en sjelden immunmediert systemsykdom som fører til inflammasjon og fibrose i affiserte organer. Pankreatikobiliær affeksjon er vanligst, men over halvparten av pasientene har multiorganaffeksjon. Grunnet variabel organaffeksjon er sykdommen en relevant differensialdiagnose ved en rekke symptombilder, og nærmest alle subspesialister kan møte slike pasienter i sin praksis. Tidlig behandling er avgjørende for å forhindre progredierende fibrose og irreversibelt tap av organfunksjon.
