Et generisk legemiddel har samme mengde virkestoff og samme eller lignende legemiddelform som et referanselegemiddel. Dokumentasjon av bioekvivalens baseres på sammenligning av farmakokinetiske egenskaper i en liten gruppe forsøkspersoner.
For generiske legemidler er det ikke nødvendig å dokumentere effekt og sikkerhet. Beslutning om markedsføringstillatelse baseres på biologisk tilgjengelighet av virkestoffet, og forskjellige farmakokinetiske variabler sammenlignes mellom det generiske legemidlet (testprodukt) og et referanseprodukt. De to viktigste variablene man benytter for å vise «likhet» eller bioekvivalens, er arealet under plasmakonsentrasjonskurven (area under the curve, AUC) og maksimal plasmakonsentrasjon (Cmaks). Dersom tiden fra inntak til maksimal plasmakonsentrasjon (Tmaks) anses å ha klinisk betydning, sammenlignes også denne (1).
Krav til dokumentasjon
Man kan ikke vise at to produkter er identiske, og det er essensielt å definere hvor store avvik som skal kunne aksepteres for at to produkter skal kunne anses som bioekvivalente. Hovedregelen er at et 90 %-konfidensintervall for gjennomsnittlig differanse mellom test- og referanseprodukt for logaritmetransformerte verdier av henholdsvis AUC og Cmaks må være innenfor ± 20 % (1). Dette svarer til at man ønsker å sikre at sann gjennomsnittlig ratio med høy sannsynlighet ligger innenfor området 0,80–1,25. Den faste definisjonen av bioekvivalens er altså 0,80 < AUCtest / AUCreferanse < 1,25 og tilsvarende for Cmaks. Akseptanseområdet kan synes relativt vidt, og de valgte grensene må ses i sammenheng med forventet variasjon i absorpsjon, distribusjon, metabolisme og eliminasjon både mellom og innen individer.
Figur 1 viser fem forskjellige eksempler på 90 %-konfidensintervall for gjennomsnittlig forhold mellom AUC for test- og referanseprodukt. I eksempel 3 og 4 er produktene bioekvivalente fordi hele intervallet ligger innenfor området 0,80–1,25. Bioekvivalens anses vist også når det er påvist statistisk signifikant forskjell i AUC, slik som i eksempel 4.
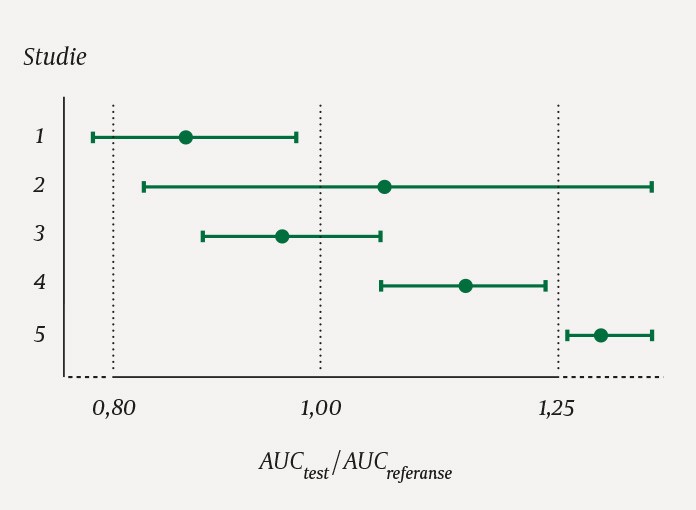
Figur 1 Tenkte resultater av fem forskjellige bioekvivalensstudier. Estimert gjennomsnittsratio for hver studie samt 90 %-konfidensintervall er skissert. I studie 3 og 4 er produktene bioekvivalente fordi hele intervallet ligger innenfor området 0,80–1,25.
Forsøksplan og statistisk analyse
Bioekvivalensstudier utføres vanligvis med friske, frivillige forsøkspersoner. Forsøkene er alltid overkrysningsforsøk, det vil si at alle forsøkspersonene får både testprodukt og referanseprodukt. Hver forsøksperson sammenlignes altså med seg selv. Det er vanlig å inkludere minst 12 individer, men kun for legemidler med høy variasjon i farmakokinetikk inkluderes det normalt mer enn 30 forsøkspersoner. Forsøket består som regel av to perioder, og forsøkspersonene får produktene i tilfeldig rekkefølge (sekvens). Den intraindividuelle variasjonen for hvert av produktene kan altså vanligvis ikke estimeres.
Hver plasmakonsentrasjonsprofil oppsummeres med AUC og Cmaks (og eventuelt Tmaks). Gjennomsnittlig differanse mellom logaritmetransformerte verdier for de to produktene (test og referanse) estimeres ofte ved variansanalyse (ANOVA) der man justerer for eventuelle periode- og sekvenseffekter. Et 90 %-konfidensintervall for gjennomsnittlig differanse estimeres også, og resultatene transformeres tilbake til opprinnelig skala. En såkalt periodeeffekt kan oppstå hvis det er en systematisk forskjell mellom resultatene for de forskjellige tidspunktene legemidlene er testet på. En sekvenseffekt vil oppstå hvis det er forskjell mellom personer som får referanseprodukt først og deretter testprodukt, og personer som får produktene i omvendt rekkefølge. Det siste unngås ved at man randomiserer hvilke forsøkspersoner som skal få hvilket produkt først. Dersom den statistiske analysen viser klare periode- eller sekvenseffekter, vil det være naturlig å sette spørsmålstegn ved kvaliteten på forsøket.
Et eksempel
Tabell 1 viser resultater fra en tenkt biotilgjengelighetsstudie. AUC avviker betraktelig for testprodukt sammenlignet med referanseprodukt for enkelte forsøkspersoner, for eksempel nummer 10 og 11. Variasjonen mellom individer er også stor. Dette er vanlig når man skal dokumentere bioekvivalens.
Tabell 1
AUC-verdier for tolv forsøkspersoner i en tenkt studie av biologisk tilgjengelighet.
|
Forsøksperson
|
AUCtest
(ng/ml∙t)
|
AUCreferanse
(ng/ml∙t)
|
|
1
|
171
|
169
|
|
2
|
196
|
200
|
|
3
|
241
|
259
|
|
4
|
188
|
187
|
|
5
|
169
|
160
|
|
6
|
171
|
218
|
|
7
|
127
|
117
|
|
8
|
136
|
173
|
|
9
|
132
|
115
|
|
10
|
135
|
228
|
|
11
|
195
|
241
|
|
12
|
376
|
342
|
I eksemplet blir gjennomsnittsratioen estimert til 0,93. Det ser altså ut til at AUC i gjennomsnitt er litt lavere for det nye produktet enn for referansen. Et 90 %-konfidensintervall blir estimert til 0,84–1,02. Fordi dette konfidensintervallet ligger innenfor grensene 0,80 og 1,25, vil de to produktene bli ansett som bioekvivalente.
Fortolkning
At et 90 %-konfidensintervall for gjennomsnittlig AUC-ratio ligger innenfor de fastsatte grensene 0,80 og 1,25, betyr ikke nødvendigvis at forholdet mellom de to produktene ligger mellom 0,80 og 1,25 for alle enkeltindivider. Som i kliniske studier av effekt og sikkerhet trekker vi slutninger om gjennomsnittet i en populasjon, ikke om effekten for en enkelt forsøksperson eller pasient. Kanskje kan pasienten ha rett hvis han hevder at effekten av legemidlet ikke er like god som før, eller at han har fått bivirkninger etter at apoteket har byttet til rimeligste alternativ?
