Diagnostikken av pasienter med utviklingsavvik og dysmorfe trekk har tradisjonelt vært basert på en skjønnsmessig klinisk vurdering, De fleste medfødte syndromer er svært sjeldne, og variasjonsbredden i fenotype ved den enkelte tilstand kan være stor. Mange pasienter får derfor kun en deskriptiv diagnose som ikke alltid er til hjelp med tanke på prognose, terapi og gjentakelsesrisiko.
Noonans syndrom, Costellos syndrom og kardiofaciokutant (CFC) syndrom er en gruppe tilstander kjennetegnet av kortvoksthet, forsinket motorisk utvikling, lærevansker/psykisk utviklingshemning, hjertefeil, hudforandringer og dysmorfe trekk. Mens Noonans syndrom er en velkjent klinisk enhet (1, 2), er erkjennelsen av Costellos syndrom (3, 4) og kardiofaciokutant syndrom (5, 6) av nyere dato. Noonans syndrom regnes som et av de vanligste medfødte syndromer med anslått forekomst på 1:2000 levendefødte. Kardiofaciokutant syndrom og Costellos syndrom er betydelig sjeldnere tilstander.
Gjennombruddet i molekylærgenetisk diagnostikk av denne syndromgruppen var påvisningen av at mutasjoner i genet PTPN11 forårsaker om lag halvparten av alle tilfeller av Noonans syndrom (7). PTPN11 koder for proteinet SHP2, en tyrosinfosfatase som deltar i intracellulær signaloverføring (fig 1). SHP2 påvirker RAS-proteiner. Disse utgjør et knutepunkt i vekstregulering ved å overføre stimuli fra celleoverflaten til kjernen via en kaskade av fosforyleringer. Denne såkalte mitogenaktivert proteinkinase (MAPK)-signalveien har vært mye studert, ikke minst fordi den er overaktivert ved flere typer kreft (8). Ofte skyldes overaktiveringen at svulstenes DNA har ervervet en mutasjon i ett av de tre RAS-genene, benevnt HRAS, KRAS og NRAS. Det aktuelle genet er da endret bare i tumorcellene, og mutasjonene kalles derfor somatiske. Dette er i motsetning til konstitusjonelle mutasjoner som man er født med og som vanligvis finnes i alle kroppens celler.

Figur 1 Oversikt over RAS/MAPK-signalveien og assosierte syndromer. Vekstfaktorer bindes til reseptorer i cellemembranen og overfører signaler til cellekjernen via en kaskade av proteiner. I kjernen aktiveres bestemte genprogrammer, som resulterer i celledeling eller differensiering. RAS-proteinene fungerer som sentrale, molekylære «brytere» i signalkaskaden. De veksler mellom en GTP-bundet, aktiv form og en GDP-bundet, inaktiv tilstand. Medfødte mutasjoner er funnet i genene som koder for proteinene, vist med blå farge. Mutasjoner i genene HRAS og KRAS gjør at RAS-proteinene rammes direkte og blir overaktive. Ved mutasjoner i genene PTPN11 (SHP2) og SOS1 er det signaloverføringen fra membranreseptorene til RAS som blir forstyrret. Andre mutasjoner (i genene BRAF, RAF1, MEK1 og MEK2) fører til at signalet fra RAS og videre inn i kjernen blir konstant påskrudd. Proteiner i MAPK-signalveien hvor man hittil ikke har påvist mutasjoner i de tilhørende genene, er vist som åpne sirkler. LEOPARD-syndrom er en tilstand nær beslektet med, men likevel distinkt forskjellig fra Noonans syndrom. De andre syndromene er beskrevet i teksten
Costellos syndrom viser mange fenotypiske likhetstrekk med Noonans syndrom, men pasientene har økt risiko for å utvikle kreft. Dette førte til oppdagelsen av at syndromet skyldes medfødte mutasjoner i HRAS (9). Store materialer av syndrompasienter ble deretter systematisk undersøkt for mutasjoner i andre gener i MAPK-signalveien. Slik fant man at Noonans syndrom kan forårsakes av medfødte mutasjoner i genene KRAS, RAF1 og SOS1, mens kardiofaciokutant syndrom skyldes mutasjoner i KRAS, BRAF eller i gener som koder for MEK-proteiner (fig 1).
Klinisk er det glidende overganger mellom syndromene slik at det for en gitt pasient kan bli nødvendig å få undersøkt en rekke gener i MAPK-signalveien. Den mest entydige sammenhengen mellom genotype og fenotype er etablert for HRAS-genet. Pasienter med klinisk sikker Costello-diagnose har som regel mutasjon i HRAS. For en oppsummering av klinisk bilde og molekylærgenetikk ved syndromer i MAPK-signalveien anbefales to oversiktsartikler (10, 11), en ny bok (12) og de fritt tilgjengelige oversiktsartiklene betegnet GeneReviews på nettstedet www.genetests.org.
Molekylærgenetisk diagnostikk av medfødte forstyrrelser i MAPK-signalveien er et felt som har beveget seg fra forskning til klinisk praksis på få år. Her vil vi illustrere dette ved å beskrive to pasienter. Videre rapporterer vi våre erfaringer med DNA-undersøkelse i et norsk materiale av pasienter i denne syndromgruppen.
Materiale og metode
Vi tilbød ved Gades institutt i 2006–07 mutasjonsanalyse av de fire genene HRAS, KRAS, BRAF og RAF1. Pasienter som var aktuelle for genanalysen, skulle primært ha Costellos syndrom eller kardiofaciokutant syndrom som mulig klinisk diagnose. Videre undersøkte vi pasienter med mistenkt Noonans syndrom som hadde negativt testresultat med henblikk på mutasjoner i PTPN11-genet ved DNA-sekvensering. Også pasienter uten klinisk diagnose, men med et medfødt syndrom karakterisert ved kortvoksthet, dysmorfe trekk og forsinket psykomotorisk utvikling (eventuelt med hjertefeil i tillegg) ble inkludert.
Vi fikk tilsendt blod- eller DNA-prøver fra medisinsk-genetiske avdelinger og barneavdelinger fra hele landet, i alt fra 23 pasienter. DNA ble ekstrahert fra EDTA-blod ved standardmetoder, og eksoner fra de aktuelle genene ble mangfoldiggjort ved hjelp av polymerasekjedereaksjon og sekvensert. For HRAS og KRAS ble hele den kodende regionen undersøkt, mens det for BRAF og RAF1 bare ble sekvensert eksoner hvor internasjonale rapporter tidligere hadde beskrevet mutasjonsfunn. DNA-analysene ble vanligvis gjort i rekkefølgen HRAS-KRAS-RAF1-BRAF. Ved positivt funn i et gen ble det ikke gjort flere molekylærgenetiske analyser. Fire av pasientene er rapportert tidligere (13, 14).
Resultater
Pasient 1. Denne gutten (fig 2a) var første barn av friske, ubeslektede foreldre. Svangerskapet forløp normalt, men ultralyd hadde vist at fosteret var stort for svangerskapsalderen. Han ble født ved spontan vaginal fødsel i 29. svangerskapsuke. Fødselsvekten (1 405 g) var over 50-prosentilen korrigert for prematuritet, lengden (40,5 cm) var under 50-prosentilen, mens hodeomkretsen (29 cm) var under 97,5-prosentilen. Ved undersøkelse sju måneder gammel hadde gutten hypertelorisme, redusert bitemporal avstand, flat og bred neserot, lavtsittende bakoverroterte ører og litt grove ansiktstrekk. Hendene var kraftige. Han hadde rikelig med hud på hender og føtter, samt dype furer i håndflatene (fig 2b).
Som nyfødt utviklet pasienten en behandlingsresistent, symptomgivende kaotisk atrietakykardi. Ernæringsvansker gjorde at han måtte sondemates de første månedene. Ved ti måneders alder var hans psykomotoriske utvikling generelt forsinket. Han hadde dårlig hodekontroll, kunne ikke sitte uten støtte og brukte hendene lite. Ved kardiologisk kontroll 20 måneder gammel var han symptomfri og har ikke hatt behov for hjertemedisiner siden. Gutten gikk like før han var tre år gammel. Ved undersøkelse 3,5 år gammel var han sosial og blid, med et ekspressivt ordforråd på ca. 20 ord. Høyden var under 2,5-prosentilen, vekt var på 2,5-prosentilen, mens hodeomkretsen var på 75-prosentilen. I tillegg til å være liten av vekst med relativ makrokefali hadde han prominent panne, hypertelorisme, fyldig underleppe og litt grove ansiktstrekk. Hendene var forholdsvis store med rikelig med hud.
Kombinasjonen av sykehistorie og funn ved klinisk undersøkelse sju måneder gammel vakte mistanke om Costellos syndrom. Diagnosen ble verifisert ved påvisning av mutasjonen Gly12Ser i HRAS-genet (fig 2c). Mutasjonen kunne ikke påvises i blodprøver fra foreldrene og ble derfor ansett som nyoppstått.
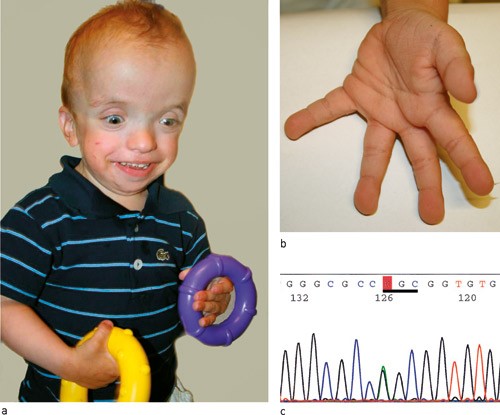
Figur 2 a) Pasient 1 med Costellos syndrom og mutasjon i HRAS-genet, alder tre år og ni måneder. b) Pasientens hånd med rikelig hud og dype furer i håndflaten. Bildene er gjengitt med tillatelse fra foreldrene. c) Den heterozygote mutasjonen c.34G > A som fører til aminosyreendringen Gly12Ser i HRAS-proteinet. Kodon 12 er understreket
Pasient 2. Denne gutten (fig 3a) var tredje barn av friske, ubeslektede foreldre. Slektsanamnesen var positiv for hypertrofisk kardiomyopati. Svangerskapet var komplisert av betydelig polyhydramnion. Han ble født ved spontan vaginal fødsel i 35. svangerskapsuke. Fødselsvekten (2 750 g) var på 50-prosentilen korrigert for prematuritet, lengden (45 cm) var under 50-prosentilen, mens hodeomkretsen (37 cm) var over 97,5-prosentilen. Gutten hadde andre levedøgn forbigående takykardi. Hypertrofisk kardiomyopati, atrieseptumdefekt og perifere pulmonalstenoser ble diagnostisert. Han ble skrevet ut åtte uker gammel med betablokkerbehandling og uten tegn til hjertesvikt. Sykehistorien og utseendet gjorde at det ble rekvirert mutasjonsanalyse av PTPN11-genet. Denne var negativ.
Fire måneder gammel var gutten alvorlig syk med pneumoni og hjertesvikt og ble re-innlagt. Lengde (54 cm) og vekt (4,8 kg) var like under 2,5-prosentilen, mens hodeomkretsen (42 cm) var på 75-prosentilen. Han hadde en prominent kvadratisk panne med redusert bitemporal avstand, periorbital hevelse, fremtredende lyseblå øyne, lyse sparsomme øyebryn, fyldige kinn, kort nese, bred neserot, kort nakke, lateralt plasserte brystvorter og oppløftede øreflipper (fig 3a). Videre hadde han «overflødig» hud i nakke og over håndrygg (fig 3b) med dype furer i håndflatene.
Et par uker senere døde pasienten av sin behandlingsresistente hjertesykdom. Ved obduksjon ble det funnet hypertrofisk høyre og venstre ventrikkel, valvulær pulmonalstenose, vid truncus pulmonalis, stor atrieseptumdefekt, vide lungevener på venstre side og lunger med stuvingstegn. Diagnosen Noonans syndrom med hypertrofisk kardiomyopati ble verifisert i oppbevart blodprøve post mortem ved funn av mutasjonen Ser257Leu i RAF1-genet (fig 3c). Mutasjonen kunne ikke påvises i blodprøver fra foreldrene og ble derfor ansett som nyoppstått.
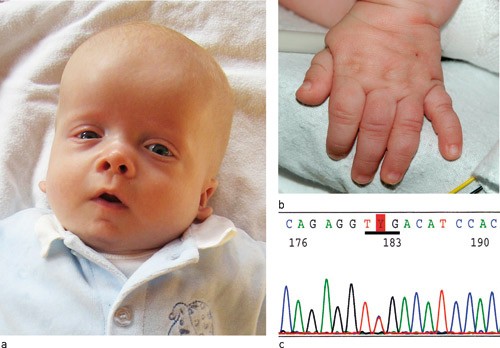
Figur 3 a) Pasient 2 med Noonans syndrom, komplisert medfødt hjertefeil inkludert hypertrofisk kardiomyopati og mutasjon i RAF1-genet, alder 3,5 måneder. b) Pasientens hånd med løs, «overflødig» hud dorsalt. Bildene er gjengitt med tillatelse fra foreldrene. c) Den heterozygote mutasjonen c.770C > T som fører til aminosyreendringen Ser257Leu i RAF1-proteinet. Kodon 257 er understreket
Mutasjonsfunn i hele pasientmaterialet
Av de 23 undersøkte pasientene, var to voksne (13) og to tenåringer. Resten var barn født i perioden 1998–2007. Hos ti av pasientene (43 %) ble det gjort mutasjonsfunn. Disse pasientene var alle heterozygote for de påviste genforandringene, noe som samsvarer med at de aktuelle tilstandene skyldes dominante mutasjoner. Det vanligste funnet var en mutasjon i kodon 12 eller 13 i HRAS-genet (fem pasienter), hvorav tre hadde Gly12Ser som er den hyppigste mutasjonen ved Costellos syndrom. Tre pasienter hadde mutasjoner i KRAS-genet og to i RAF1-genet. For åtte av de ti pasientene med mutasjonsfunn var foreldre-DNA tilgjengelig. Foreldreprøvene var i alle åtte tilfeller negative for barnets mutasjon.
Diskusjon
De to beskrevne pasientene illustrerer at den diagnostiske syndromutredningen fortsatt er fundamentert i kliniske manifestasjoner. Nye molekylærgenetiske muligheter gjør imidlertid syndromdiagnostikken mer presis og er noen ganger helt avgjørende for å skille mellom ulike differensialdiagnoser. Pasient 1 hadde mange kjennetegn på Costellos syndrom (høy fødselsvekt, ernæringsvansker, postnatal vekstretardasjon, generell utviklingsforsinkelse, blid og sosial personlighet, dysmorfe ansiktstrekk, «overflødig» hud, hjertesykdom). Den kliniske diagnosen ble bekreftet ved funn av mutasjon i HRAS-genet.
Pasient 2 var betydelig mer problematisk å klassifisere klinisk, og molekylærgenetisk testing ble avgjørende for diagnosen. Uttalt polyhydramnion er en vanlig svangerskapskomplikasjon ved både kardiofaciokutant syndrom og Noonans syndrom. Alvorlig kardiomyopati er typisk for Noonans syndrom forårsaket av mutasjoner i RAF1-genet (15), mens mutasjoner i PTPN11 oftere blir påvist hos pasientene med pulmonal stenose uten kardiomyopati (7). Hudforandringene og ansiktstrekkene til pasienten vakte mistanke om kardiofaciokutant syndrom. Den endelige diagnosen ble fastslått ved funn av mutasjonen Ser257Leu i RAF1-genet, en genfeil tidligere påvist hos flere pasienter med Noonans syndrom og alvorlig/letal hypertrofisk kardiomyopati (15). Analyse av blodprøver fra foreldrene indikerte at mutasjonen hos barnet var nyoppstått, noe som gjorde det rimelig å konkludere med at hans hypertrofiske kardiomyopati var uten sammenheng med kardiomyopati tidligere påvist hos slektninger.
Funn av mutasjoner i MAPK-signalveien kan være viktig for pasientoppfølgingen. Barn med disse syndromene har tendens til alvorlige spiseproblemer, og sondemating vil ofte være påkrevd i første leveår. Videre vil mange ha hjertefeil som krever spesiell overvåking. Personer med PTPN11-mutasjon kan ha økt blødningstendens. Hos enkelte pasienter, særlig dem med kardiofaciokutant syndrom, kan moderat til alvorlig utviklingshemning forventes. Situasjonen er annerledes ved Noonans syndrom: Om lag en tredel av pasientene vil være mildt rammet, eventuelt bare ha lærevansker. Behandling med veksthormon kan være aktuelt hos barn med Noonans syndrom (16).
Et annet viktig aspekt er kreftrisiko siden MAPK-signalveien ofte er overaktivert i ondartede svulster (8). Samtlige gener her kan i prinsippet tenkes å være involvert i kreftutvikling. KRAS er for eksempel hyppig mutert i bukspyttkjertelkreft og tykktarmskreft. De somatiske KRAS-mutasjonene i svulster er imidlertid helt forskjellige fra de konstitusjonelle mutasjonene man har funnet ved syndromer i MAPK-signalveien. Inntil vi har fått mer erfaring med pasientgruppen, antas det derfor at personer med medfødte KRAS-mutasjoner ikke har forhøyet kreftrisiko. Ved Costellos syndrom, derimot, forekommer de medfødte HRAS-mutasjonene i de posisjonene som er somatisk endret i mange svulster (kodon 12 og 13). Det er vist at pasienter med Costellos syndrom har forhøyet risiko for kreftformer som rabdomyosarkom og nevroblastom i barnealder og for blærekreft i voksen alder (17, 18). En av pasientene i vår serie ble diagnostisert med blærekreft da hun var 44 år gammel (13).
Påvisning av mutasjon hos et affisert barn vil naturlig nok føre til spørsmål fra foreldrene om gjentakelsesrisiko i eventuelle påfølgende svangerskap. Vi fant, i tråd med erfaringen internasjonalt, at medfødte, patogene forandringer i RAS- og RAF-gener er nyoppstått. Dette vil si at mutasjonene har kommet til enten under danningen av foreldrenes gonader/kjønnsceller eller kort tid etter befruktningen. Gjentakelsesrisikoen for søsken av en affisert person vil derfor være svært liten. Fosterdiagnostikk for mutasjoner i RAS/RAF-gener vil som hovedregel ikke være aktuelt i familier som har fått et barn med kardiofaciokutant syndrom eller Costellos syndrom. Gonademosaikk kan imidlertid tenkes å forekomme. Vi kan derfor ikke helt utelukke at gjentakelsesrisikoen for søsken kan være økt i noen få familier som alt har et affisert barn. Men vi har så langt ikke mulighet til på forhånd å identifisere eventuelle familier med forhøyet gjentakelsesrisiko for kardiofaciokutant syndrom eller Costellos syndrom. De to søsknene som Søvik og medarbeidere har beskrevet (13), må anses som et helt eksepsjonelt sammentreff av to forskjellige mutasjonsbegivenheter i en og samme familie.
Også mange tilfeller av Noonans syndrom skyldes nyoppståtte mutasjoner, men det finnes familier med affiserte personer i flere generasjoner. Fenotypen er svært variabel, selv innenfor samme familie. Hvis mor eller far har Noonans syndrom, er det i hvert svangerskap 50 % sannsynlighet for at fosteret arver genfeilen. Når familiens mutasjon er kjent, er det teknisk mulig å tilby målrettet fosterdiagnostikk. Etter dagens praksis vil en søknad om fosterdiagnostikk grunnet økt risiko for Noonans syndrom bli innvilget.
Vårt materiale er trolig mest komplett for perioden 2003–07. Her fant vi sju pasienter med RAS- eller RAF-mutasjon. Dette kan tyde på en gjennomsnittlig forekomst på 1–2 tilfeller i året i Norge, noe vi anser for å være et minimumsestimat. Det ble fra vår side ikke gjort forsøk på komplett innhenting av norske pasienter, og antageligvis visste ikke alle aktuelle sykehusavdelinger om tilbudet om gentesting. Videre ble ikke gener som SOS1, MEK1 og MEK2 testet i vår undersøkelse. Dette er alle gener hvor mutasjoner er funnet i syndromgruppen (fig 1). Det er derfor grunn til å tro at det her i landet finnes en rekke udiagnostiserte syndrompasienter med mutasjoner i RAS/MAPK-signalveien. Selv Noonans syndrom, den mest kjente tilstanden i diagnosegruppen, er antageligvis underdiagnostisert. Vi vil fremheve at en årsaksdiagnose kan være av stor verdi for familien, selv når pasienten er langt opp i voksen alder.
Gentesting for syndromene nevnt i denne artikkelen har beveget seg et langt skritt fremover det siste tiåret. Den molekylærgenetiske utredningen krever systematisk analyse av mange ulike gener, og laboratoriediagnostikken er i Norge samlet ved Senter for medisinsk genetikk og molekylærmedisin, Haukeland universitetssykehus. Man kan også kontakte en av landets medisinsk-genetiske avdelinger for å diskutere den diagnostiske utredningen av pasienten eller for henvisning til klinisk genetiker.
Molekylærgenetisk utredning av syndromer med affisert RAS/MAPK-kaskade har i dag klar diagnostisk og oppfølgingsmessig verdi. I fremtiden kan det tenkes at det utvikles medikamenter som hemmer overaktiviteten i denne signalveien (19). I så fall vil laboratorieanalysene bli en absolutt forutsetning for rett pasienthåndtering.
