For lite eller for mye kromosommateriale er den vanligste årsaken til medfødte utviklingsavvik. Kromosomfeil er siden 1960-årene blitt påvist ved lysmikroskopi, som er en nokså grov undersøkelse. I 2005 ble DNA-matrisebaserte metoder tatt i klinisk bruk, og man kan nå påvise kromosomavvik ned til enkeltgennivå. Slik molekylær karyotyping gir presis informasjon om hvilke gener som foreligger i endret kopitall, men tolkingen kan være vanskelig og forutsetter gode kliniske opplysninger.
Mennesket har 23 kromosompar og to kopier av de fleste gener. Når et kromosomområde er duplisert, vil genene i denne regionen foreligge i tre kopier istedenfor to. Ved delesjoner reduseres kopitallet fra to til en. Slik genomisk ubalanse er den langt vanligste påvisbare årsak til medfødte syndromer med utviklingsavvik og dysmorfe trekk med eller uten organmisdannelser.
Lysmikroskopisk undersøkelse av trypsinbehandlede og Giemsa-fargede kromosomer fra celler i delingsfase, såkalt G-bånd-karyotyping (fig 1), har vært standard cytogenetisk analysemetode siden 1960-årene, og er fremdeles rutinemetoden for å lete etter kromosomfeil. Denne analysen krever dyrking av celler og er tid- og ressurskrevende. I tillegg blir mange kromosomfeil ikke oppdaget med denne grove metoden fordi de er for små til å være synlige i mikroskopet (< 5 – 15 millioner basepar (Mb)).
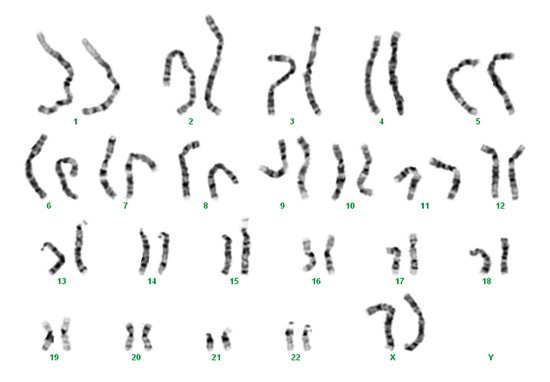
Figur 1 G-bånd-karyotyping (trypsinbehandlede og Giemsa-fargede metafasekromosomer) hos en normal kvinne med karyotype 46,XX
For å påvise submikroskopiske (også kalt kryptiske) kromosomavvik er det i løpet av de siste 10 – 15 år utviklet supplerende metoder som fluorescerende in situ-hybridisering (FISH) og kromosombasert høyresolusjons komparativ genomisk hybridisering (HR-CGH) (1, 2). FISH-analyser er målrettede undersøkelser som benyttes når det er klinisk mistanke om spesifikke kromosomavvik. Det benyttes en fluorescensmerket DNA-bit (probe) som binder seg spesifikt til den komplementære DNA-sekvensen i den aktuelle kromosomregionen hos pasienten. Bindingsprosessen kalles hybridisering. Et velkjent eksempel er FISH-analyse for 22q11.2-delesjonen, som er årsak til DiGeorges syndrom (fig 2). Et alternativ til FISH-analyse er MLPA-analyse (multipleks ligeringsavhengig probeamplifikasjon) (e-fig 3). I motsetning til FISH-analyser som utføres på kromosomer fra levende celler, krever MLPA-analysen kun renset DNA. Ved både FISH- og MLPA-analyse blir et begrenset antall forhåndsdefinerte kromosomregioner undersøkt. HR-CGH-analyse er derimot en generell screeningmetode for genomisk ubalanse, men sensitiviteten er bare dobbelt så høy som ved vanlig lysmikroskopisk kromosomundersøkelse (3).
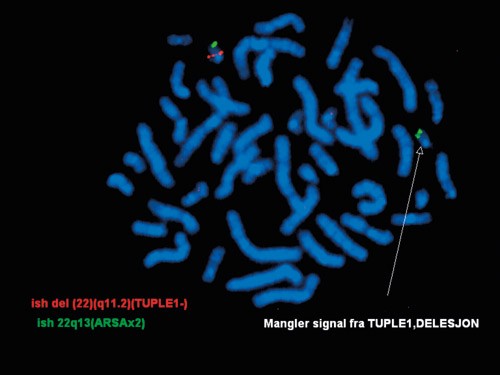
Figur 2 FISH-analyse som viser en delesjon av et område på kromosom 22 forbundet med DiGeorges syndrom. Delesjonen identifiseres ved at man ser grønne kontrollsignal svarende til et annet kromosom 22-område på begge kromosom 22, men bare ett rødt signal fra 22q11.2 området, der delesjonen forekommer
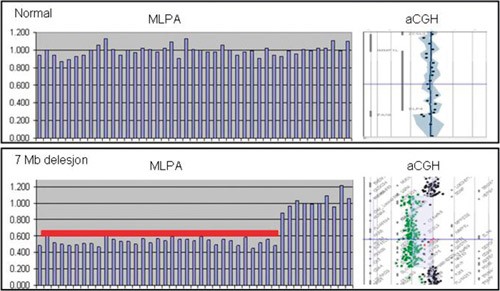
Figur 3 En sammenlikning av resultater fra MLPA-analyse (multipleks ligeringsavhengig probeamplifikasjon) og aCGH-analyse (matrisebasert komparativ genomhybridisering) hos en normal (øverst) og en pasient heterozygot for en 7 Mb-delesjon som bl.a. omfatter genene PAX6 og WT1. Tap av førstnevnte gen er årsak til aniridi, mens tap av sistnevnte gen gir økt risiko for Wilms tumor i nyrene. Denne delesjonen ville mest sannsynlig ikke bli oppdaget ved karyotyping. MLPA-analyse gir informasjon om at utvalgte gener er deletert (halvering av topphøyder avmerket i rødt), mens aCGH-analyse gir nøyaktig informasjon om hvor delesjonen begynner og slutter og hvilke gener som er i regionen
Vi beskriver her en ny type høyresolusjons molekylær kromosomanalyse, en DNA-matrisebasert metode som ofte kalles «array-CGH» (array-based comparative genomic hybridization). Molekylær karyotyping på slike DNA-matriser tilbys både ved Avdeling for medisinsk genetikk, Oslo universitetssykehus, Ullevål og ved Senter for medisinsk genetikk og molekylærmedisin, Haukeland universitetssykehus. Analysene kan detektere små delesjoner og duplikasjoner, også kalt kopitallsvarianter (copy number variations, CNV), i hele genomet med stor nøyaktighet. Siden metoden ble tatt i klinisk bruk i 2005, er det beskrevet flere nye mikrodelesjons- og mikroduplikasjonssyndromer (4). Ingen annen laboratoriemetode har tilnærmelsesvis like høy funnprosent når det gjelder å påvise årsaken til psykomotorisk utviklingshemning. Å stille en årsaksdiagnose hos et barn med et medfødt syndrom kan ha betydning for prognose, medisinsk oppfølging og vurdering av gjentakelsesrisiko for andre i slekten. Mange foreldre opplever dessuten en lettelse når endelig en årsaksdiagnose blir stilt etter en lang periode med usikkerhet.
Metoden
Metoden for å lete etter endringer i kopitall ved hjelp av DNA-matriser er en videreutvikling av HR-CGH-metoden (1). Mens man med denne metoden kan identifisere avvik ned til omkring 4 Mb, kan man med matrisebaserte CGH-undersøkelser påvise avvik på 0,1 Mb eller mindre, dvs. helt ned på enkeltgennivå.
Krav til det biologiske materialet er 1 – 5 ml EDTA-blod eller ca. 1 µg genomisk DNA isolert fra dyrkede celler, vev eller munnslimhinne. Prinsippet for metoden som benyttes ved Oslo universitetssykehus, Ullevål, er skissert i figur 4. Her benytter man matriser med opptil 244 000 oligonukleotider, noe som gjør at man i teorien kan påvise delesjoner eller duplikasjoner ned til 0,005 Mb (5 kb). Om kort tid vil vi ha muligheten til å analysere 1 million oligonukleotider samtidig. Ved Haukeland universitetssykehus benyttes en matrise med over 1,8 millioner oligonukleotider (fig 5). Den matrisen inneholder også oligonukleotider med kjente enkeltnukleotidpolymorfismer (enkeltbasevariasjon, single nucleotide polymorphism, SNP) og kan derfor også brukes til å påvise homozygositetsområder og uniparental isodisomi. Slike homozygositetsområder kan noen ganger indikere det kromosomale locus (genområdet) for en recessivt arvelig tilstand. Funn av flere store homozygositetsområder tyder på at foreldrene er nært beslektet. Selv om den kopitallsanalysen som inkluderer enkeltnukleotidpolymorfismer teknisk sett er noe annerledes enn den som anvendes ved Oslo universitetssykehus, Ullevål, gir begge metoder samme type resultat når det gjelder kopitallsvariasjon, og sensitiviteten er sammenliknbar. For enkelhets skyld vil vi i det følgende bruke aCGH-analyse som et fellesnavn for begge metodene.
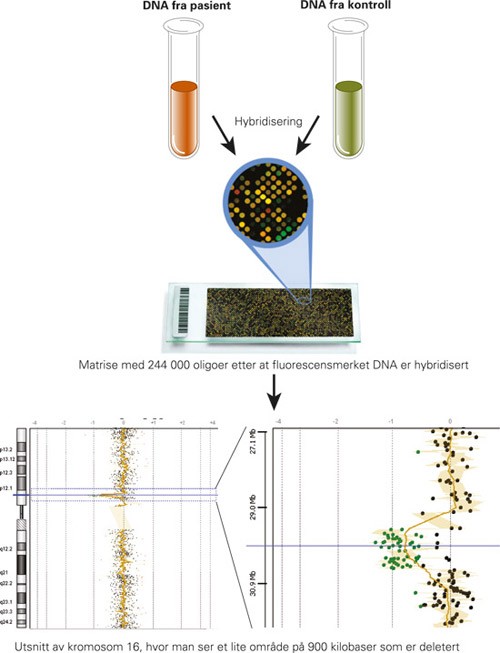
Figur 4 Ved matrisebasert komparativ genomhybridisering (aCGH) renses DNA fra en blod- eller vevsprøve, og etter oppkutting merkes DNA-fragmentene med rød fluorescens. Samtidig merkes DNA fra en kontrollperson med grønn fluorescens. Prøvene blandes og legges på en 2 x 6 cm stor glassbrikke (matrise, også kalt array) der korte syntetiske DNA-molekyler kalt oligonukleotider (oligoer) sitter tett i tett i definerte posisjoner. Alle oligoene på matrisen har kjent DNA-sekvens – vi vet med andre ord hvilke gener/områder av genomet de representerer. Ved hybridisering vil DNA-bitene binde seg til de oligoene som representerer komplementær DNA-sekvens. Etter at DNA er bundet, undersøkes glassbrikken med en laserscanner, og fluorescenssignalene fra alle posisjonene analyseres med avanserte dataverktøy. Når den relative signalstyrken i hver posisjon plottes i forhold til hvor på kromosomene DNA-sekvensen er hentet fra, klarer man å identifisere områder i genomet der pasienten har avvikende kopitall. Grønne posisjoner indikerer at den DNA-sekvensen som oligoen detekterer, er underrepresentert (deletert) i prøven sammenliknet med kontrollen, mens røde posisjoner indikerer at det er for mye DNA til stede (duplikasjon). Gule posisjoner betyr at det er samme kopitall i de to prøvene
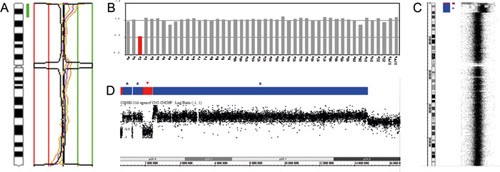
Figur 5 Ved kromosombasert HR-CGH-analyse av DNA fra en pasient med klinisk mistanke om et sjeldent syndrom ble en duplikasjon (merket grønn) av enden på den korte armen av kromosom 2 påvist (A), mens MLPA-analyse viste det motsatte, nemlig en delesjon (rød søyle) av terminale 2p (B). Kompleksiteten av det kromosomale rearrangementet kom først frem når matrisebasert kromosomanalyse (Affymetrix 6.0 SNP-chip) ble benyttet. Her fant vi alternerende delesjoner (røde) og duplikasjoner (blå). I panel C vises hele kromosom 2, mens i panel D er det affiserte området på enden av kromosom 2 forstørret
Praktiske anvendelser
Avklaring av kjente kromosomfeil
Ved markørkromosomer og nyoppståtte translokasjoner er aCGH-analyse et meget nyttig verktøy for å forstå den kliniske betydningen av avvikene. Et markørkromosom er en liten ekstra kromosombit av ukjent opprinnelse (et 47. kromosom). Et markørkromosom kan være uten fenotypisk effekt (et tilfeldig funn) eller årsak til utviklingsavvik. En aCGH-analyse vil raskt kunne avklare hvilket kromosom markørkromosomet stammer fra, og eventuelt hvilke gener det inneholder. Translokasjoner er ombytting av kromosombiter, som regel mellom to ulike kromosomer (resiproke translokasjoner). Metoden kan ofte bringe på det rene om en translokasjon som i mikroskopet ser balansert ut, virkelig er det, eller om genetisk materiale har gått tapt i bruddstedene eller andre steder (5). Denne avklaringen er nødvendig for å vurdere om en translokasjon er klinisk signifikant eller kun representerer et tilfeldig funn. Problemstillingen er særlig aktuell ved nyoppståtte translokasjoner (3, 6). Likeledes kan aCGH-analyse benyttes til mer nøyaktig karakterisering av kjente delesjoner og duplikasjoner.
Påvisning av nye kromosomfeil
Størst nytte av aCGH-analyse har vi hos pasienter som ikke har fått påvist kromosomavvik ved hjelp av tradisjonelle kromosomundersøkelser til tross for at det kliniske bildet er vel forenlig med en kromosomfeil. Kryptiske kromosomavvik har vist seg å være omtrent tre ganger vanligere enn avvik som kan påvises i mikroskopet (3). Når aCGH-analysen har påvist avvikets posisjon og type, kan man imidlertid ofte også se avviket i mikroskopet – selv om det er blitt oversett ved tidligere lysmikroskopisk screening.
aCGH-analyse kan også påvise mosaisisme, dvs. kromosomavvik som kun foreligger i en viss andel av kroppens celler. Ved slike kromosomale mosaikktilstander finner man ofte normal karyotype ved rutinemessig analyse av T-lymfocytter dyrket fra en blodprøve. Dette skyldes at celler med kromosomfeil som regel vokser dårligere enn normale celler. Ved aCGH-analyse undersøkes DNA fra udyrkede celler, noe som muliggjør diagnostikk av mosaisisme hvis en tilstrekkelig andel av cellene (> 30 %) har ubalansert karyotype.
Indikasjoner og begrensninger
Den nye metoden påviser ikke balanserte kromosomavvik, kun genomisk kopitallsvariasjon. De fleste translokasjoner og inversjoner er balanserte, og slike balanserte avvik kan være uten fenotypiske konsekvenser utover å forårsake infertilitet og spontanaborter. Ved disse indikasjonene bør derfor dagens praksis med å rekvirere standard kromosomanalyse fortsette. Triploidi (tilstedeværelse av tre kromosomsett), som er en årsak til spontanabort, oppdages heller ikke ved aCGH-undersøkelse.
Metoden representerer en diagnostisk revolusjon som muliggjør overgang fra mikroskopisk til molekylær karyotyping (7 – 9). Den tekniske kvaliteten og sensitiviteten blir stadig bedre. Utfordringen nå er å forstå betydningen av det vi finner. Alle mennesker har kopitallsvariasjoner i genomet som bidrar til den genetiske normalvariasjonen. Variasjonen i kopitall mellom to ubeslektede funksjonsfriske personer kan samlet utgjøre 4 – 24 Mb av genomets totale ca. 3 200 Mb (10, 11). Kunnskapen om hvilke kopitallsvariasjoner som er nøytrale og hvilke som medfører risiko for sykdom eller utviklingsavvik, er mangelfull. Det er heller ikke alltid avklarende å undersøke om samme variant foreligger hos en av foreldrene eller ikke. Nyoppståtte varianter kan være uten patogenetisk betydning, og familiære varianter kan gi økt risiko for utviklingsavvik. Hvorfor visse kromosomfeil kan medføre et utviklingsavvik hos barnet, men ikke hos den av foreldrene som feilen er nedarvet fra, er stort sett ukjent.
Betydningen av genomiske avvik må alltid tolkes i lys av pasientens fenotype, og derfor er det viktig at rekvirerende lege gir gode kliniske opplysninger, dvs. opplysninger om fødselsvekt, vekst, hodeomkrets, organmisdannelser, utvikling, funksjonsnivå og eventuelle atferdsavvik. Generelt kan man si at hvis et avvik overlapper med et område eller et gen som er kjent assosiert med utviklingsavvik, er det større sannsynlighet for at kromosomfunnet er årsak til fenotypen (12). Dersom kromosomområdet er genrikt, og avviket er en delesjon på minst 0,5 Mb, er det også sannsynlig at funnet har betydning. Mindre og mer genfattige avvik som er nedarvet fra friske foreldre eller som er tidligere funnet hos friske personer, særlig duplikasjoner, representerer mest sannsynlig normalvarianter.
Enkelte kopitallsvarianter, som delesjoner i kromosombånd 15q13.3 eller duplikasjoner i kromosombånd 22q11.2, finnes hyppigere hos individer med ulike utviklingsavvik enn hos normale personer. Slike kopitallsvarianter er altså assosiert med økt risiko for utviklingsavvik. Funn av slike såkalte «sårbarhetsvarianter» kan være en tolkings- og veiledningsmessig utfordring. Man kan ofte lure på om det er kopitallsvarianten eller noe annet som er årsak til utviklingsavviket, eller kanskje kopitallsvarianten bare er en av flere medvirkende faktorer. Slike funn må tolkes med forsiktighet.
For å møte den tolkingsmessige utfordringen som aCGH-analyse representerer, kreves tett samarbeid mellom laboratorium og klinikere. Fordi mange avvik er svært sjeldne eller tilsynelatende unike, er det nødvendig at alle sikre funn blir registrert med kvalitetssikrede molekylære og kliniske data i internasjonale databaser. Slike databaser er essensielle hjelpemidler for tolking av funn. DECIPHER og ECARUCA er to Internett-baserte databaser der man kan undersøke om tilsvarende avvik er rapportert hos andre pasienter (ramme 1). Den engelske pasientforeningen Unique har utarbeidet pasientinformasjonsbrosjyrer om en rekke mer hyppig forekommende kromosomavvik, og mange familier finner disse brosjyrene nyttige. I Norge gir Frambu senter for sjeldne funksjonshemninger tilbud til familier som har et medlem med et sjeldent kromosomavvik som medfører utviklingshemning.
Vi forventer at aCGH-analyse om få år vil være den primære screeningmetode ved mistanke om medfødt kromosomfeil som gir genomisk ubalanse. Metoden vil oppdage alle kjente kromosomavvik som gir opphav til utviklingshemning, inkludert numeriske avvik (som trisomi 13, 18 og 21). Den største fordelen med aCGH-analyse er at unike og svært sjeldne kryptiske kromosomfeil også blir påvist. Samlet sett er kryptiske kromosomfeil en langt hyppigere årsak til utviklingshemning enn f.eks. trisomi 21.
Etter hvert som matrisene får høyere dekningsgrad, vil man også kunne anvende dem til å screene for monogene sykdommer. Utfordringen nå og i mange år fremover blir å forstå betydningen av mange av funnene. Inntil man har skaffet seg bedre kunnskapsgrunnlag for tolkingen av funn, må aCGH-analyse benyttes med omhu (13).
