Nevrodegenerative sykdommer hos barn er ofte autosomalt recessivt nedarvet. De fleste tilfellene er metabolske, der inngifte er en viktig risikofaktor. Diagnostikken kan være svært krevende. Tap av motoriske, kognitive og sosiale ferdigheter er vanlig. Det kreves stor innsats fra hjelpeapparatet – 15 – 30 fagpersoner kan være engasjert rundt én pasient. For et økende antall sykdommer finnes det nye og dyre behandlingsformer.
Selv om nevrodegenerative sykdommer hos barn innebærer høy mortalitet og morbiditet, er de ikke lenger uhelbredelige, da kausalt rettet behandling etter hvert er blitt tilgjengelig (1). Likevel representer disse sykdommene en stor belastning for alle involverte. En families erfaringer med påkjenningene som oppstår i forløpet av å ha et barn med nevrodegenerativ sykdom er beskrevet nedenfor.
Pasienten. Gutten, som nå er i tenårene, har hele tiden vokst lite. Han har mikrokefali, med hodeomkrets 5 cm < 2,5-prosentilen, og et «gammelmannsaktig» utseende, med uthulte øyne og lite subkutant fettvev. Høyden er 110 cm, som er 24 cm < 2,5-prosentilen og tilsvarer gjennomsnittshøyden for en femåring. Gastrostomi førte til en betydelig bedring av ernæringssituasjonen, men hadde trolig ikke tilsvarende gunstig effekt på høydetilveksten. Han satt ved åtte måneders alder og gikk med støtte 16 måneder gammel, men oppnådde ikke selvstendig gange. Hørselen begynte å svikte ved to års alder, og han fikk høreapparat. Det ble etter hvert åpenbart at han var mentalt retardert. 3 1/2 år gammel skåret han tilsvarende 16-månedersnivå på en ferdighetstest. Ved fire års alder begynte han å miste motoriske ferdigheter, og en kombinasjon av cerebellar ataksi, demyeliniserende polynevropati og spastisitet gjorde ham fullstendig avhengig av rullestol.
Gutten er i kontakt med en lang rekke spesialister – ortoped, ryggortoped, barnekirurg, øyelege, tannlege, øre-nese-hals-lege, gastroenterolog, barnenevrolog, audiopedagog, stomisykepleier og ernæringsfysiolog. Førstelinjetjenesten er involvert med bl.a. fastlege, fysioterapeut, ergoterapeut og sosionom. I tillegg kommer assistanse fra personell på skolen og avlastningshjelp for foreldrene. Samordningen av innsatsen har vært så krevende at faren mistet arbeidet på grunn av for høyt fravær for å følge opp konsultasjonene.
Situasjonen blir ikke mindre komplisert av at familien har ikke-vestlig innvandrerbakgrunn – det er utfordringer vedrørende norsk språk og kultur.
Noen år senere fødte moren en gutt til med nøyaktig samme symptomutvikling og funn, slik at familien har to barn med nevrodegenerativ tilstand – i tillegg til sine friske barn. Til tross for inngående undersøkelser på sykehus ble årsaksdiagnosen først stilt flere år etter guttene var født. Gjennombruddet kom ved bruk av internasjonale kontaktnettverk og etter relevante cerebrale MR- og CT-undersøkelser med påfølgende cellebiologiske og genetiske analyser.
Forekomst – inngifte som risikofaktor
Selv om de hver for seg er sjeldne, er nevrodegenerative sykdommer hos barn relativt hyppig forekommende samlet sett. Klinisk består de av progredierende encefalopatier (som i hovedsak rammer hjernen) og en gruppe med hovedsakelig nevromuskulære symptomer. Det finnes få studier der man har kartlagt forekomsten. I en undersøkelse utgått fra Oslo universitetssykehus (Aker og Ullevål sykehus og Rikshospitalet) fant man totalt 84 Oslo-barn med progredierende encefalopati født i årene 1985 – 2004. Til sammen representerte disse 28 forskjellige diagnoser, hovedsakelig autosomalt recessive sykdommer, men hos fire pasienter (5 %) var det X-bundet arv.
Insidensraten, som ble beregnet til 6,4 per 100 000 personår, var stabil gjennom observasjonsperioden (fig 1) (2). Den kumulative insidensen på 0,6 per 1 000 i Oslo var sammenliknbar med insidensen i en tidligere studie fra Vest-Sverige (3) og like hyppig som medfødt hydrocephalus (4) eller nevralrørsdefekter (5). Vårt materiale fra Oslo viste videre at progredierende encefalopati hos barn var sju ganger så vanlig hos barn av pakistanske foreldre som hos barn av norske (6). Forklaringen på den økte risikoen var høy forekomst av inngifte hos foreldre med bakgrunn fra Pakistan. Pakistanske foreldre som er fetter og kusine har 11 ganger så høy risiko for å få et barn med progredierende encefalopati som et par med norsk etnisk bakgrunn (fig 2) (6).
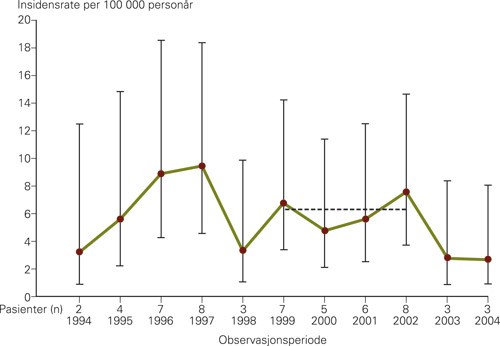
Figur 1 Insidensraten av progredierende encefalopati hos Oslo-barn i alderen 0 – 15 år var stabil i perioden 1994 – 2004 (2). 95 % konfidensintervall (KI) for hvert års insidensrate er indikert med vertikal linje. Mellom 1999 og 2002 var insidensraten 6,13 per 100 000 personår under risiko. Beregningen av personår, som var basert på antall fødsler per år, var tilnærmet riktig frem til 1999, mens antall nye sykdomstilfeller etter 2002 kan være underestimert fordi enkelte, spesielt barn født i 2003, ikke hadde rukket å utvikle sykdomssymptomer. Årene 1999 – 2002 (den stiplede horisontale linjen) var derfor den mest representative perioden for insidensberegningen
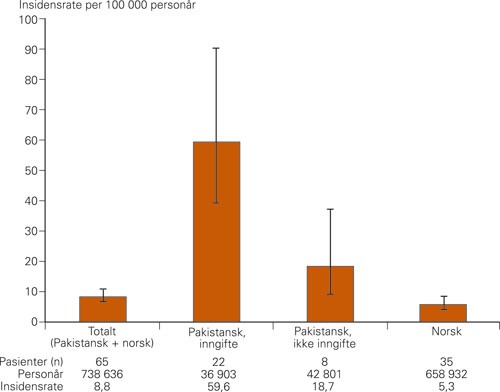
Figur 2 Insidensraten av progredierende encefalopati ble målt hos pakistanske og norske barn i henhold til foreldrenes inngiftestatus (6). Følgende insidensrateratioer kunne beregnes: barn av inngiftede foreldre med pakistansk bakgrunn (pakistansk, inngifte) versus norske barn (norsk) = 11,2; barn av inngiftede foreldre med pakistansk bakgrunn versus barn av ikke-inngiftede foreldre med pakistansk bakgrunn (pakistansk, ikke inngifte) = 3,2; barn av ikke-inngiftede foreldre med pakistansk bakgrunn versus norske barn = 3,5. Inngiftet er her definert som fetter-kusine-ekteskap. De vertikale linjene indikerer 95 % konfidensintervall. Verdiene viser at inngifte er en risikofaktor for progredierende encefalopati. I tillegg er pakistansk bakgrunn en separat risikofaktor. Årsakene til dette er komplekse og bunner blant annet i en inngiftepraksis, som er fjernere enn fetter-kusine-ekteskap, i klaner og brorskap (biraderi) i rurale strøk. Figuren er hentet fra Strømme og medarbeideres artikkel (6). © 2010, Elsevier. Gjengitt med tillatelse
I Vest-Europa og Nord-Amerika er det skepsis til inngifte, og det er forbudt ved lov i flertallet av delstatene i USA. I en oversiktsartikkel om medisinske konsekvenser av denne praksisen peker Bittles & Black (7) på at i rurale strøk der prevalensen av inngifte er 50 – 60 %, det gjelder f.eks. noen områder i Pakistan (8), oppveier de sosiale fordelene ved nært slektskap mellom foreldrene de biologiske ulempene. Et barn med medfødt autosomalt recessiv sykdom vil der kanskje ikke leve lenge fordi medisinsk behandling ikke er aktuelt på grunn av ressursmangel. I Vest-Europa og Nord-Amerika er forholdene annerledes. Der vil det være større sannsynlighet for at barnet skal leve opp, men det vil være avhengig av livslang behandling eller støtte. I Norge, i motsetning til i Storbritannia, er prevalensen av inngifte blant ikke-vestlige innvandrere synkende, og bare 16,7 % av pakistanske kvinner født i Norge er gift med sin fetter (9). Synkende prevalens av inngifte blant innvandrere på tross av økt ikke-vestlig innvandring kan forklare hvorfor insidensen av progredierende encefalopati i Oslo har holdt seg stabil (fig 1).
I alle tilfeller der det er mistanke om nevrodegenerativ sykdom, uansett inngiftestatus, henvises foreldrene til genetisk avdeling for informasjon om gjentakelsesrisikoen og mulig fosterdiagnostikk. Slik henvisning er rutine både i tilfeller der diagnosen er stilt, men også i tilfeller der årsaken er ukjent.
Symptomer og prognose
Tap av psykomotoriske ferdigheter hos barn kan være vanskelig å fange opp, spesielt i 1 – 2 års alder, da den normale utviklingen skjer raskt. Sykdommer med hovedsakelig affeksjon av hvit hjernesubstans debuterer gjerne med motoriske problemer på grunn av spastisitet. En fellesnevner for sykdommer der grå substans først blir angrepet, er epilepsi og demensutvikling. Tap av syn og hørsel og utfall fra andre organsystemer (lever, hjerte, nyre og skjelett) forekommer også ved en rekke metabolske tilstander.
Ved noen sykdommer varierer den fenotypiske utformingen med hvilken alder symptomene debuterer. Et eksempel på dette er Niemann-Picks sykdom type C (NPC), der debut i voksen alder er kjennetegnet av psykiatriske symptomer, mens samme genmutasjon gir en infantil eller tidlig juvenil form, som regel med tidlig død (10). I Oslo-studien var dødeligheten ved progredierende encefalopatier hos barn 36,9 % etter en gjennomsnittlig oppfølgingstid på 8,5 år (11). Den korteste overlevelsen ble observert hos barn med metabolsk sykdom som debuterte i neonatalperioden.
Utvidet screening?
Å stille en riktig etiologisk diagnose ved nevrodegenerative sykdommer hos barn er viktig av mange grunner. Samtidig kan det ofte være en krevende og langvarig prosess. Undersøkelse med cerebral MR-bildefremstilling (MR «imaging»; MRI) kombineres stadig oftere med MR-spektroskopi (MRS). Ved hjelp av sistnevnte undersøkelse fremstilles de vanligste metabolitter i hjernen, og den er nyttig i diagnostikken innenfor et bredt spektrum av metabolske og degenerative tilstander (12). Ett eksempel på kombinert MRI- og MRS-undersøkelse er vist i figur 3 (13). Selv etter nitid utredning med cerebral MR, biokjemiske analyser (avdeling for genetisk biokjemi, Rikshospitalet, og avdelingen for klinisk kjemi ved Sahlgrenska Universitetssjukhuset, Göteborg) og molekylærgenetiske analyser (sekvensering av ett eller flere gener eller screening av et utvalg mutasjoner) forblir en stor andel av pasientene uten etiologisk diagnose. I Oslo-studien utgjorde gruppen udiagnostiserte 20 % av pasientene, på tross av at de var blitt drøftet i et panel med utenlandske eksperter (2). Foto, video og nye digitale medier som Internett og elektroniske syndromdatabaser har imidlertid lettet det diagnostiske arbeidet vesentlig.
Stadig nye behandlingsmuligheter aktualiserer viktigheten av en presis diagnose ytterligere. Behandlingen bør settes inn før sentralnervesystemet påføres irreversibel skade. I Norge screenes i dag nyfødte for fenylketonuri og hypotyreose. Med tandemmassespektrometrimetoden (MS/MS) kan langt flere tilstander oppdages tidlig. I en del industrialiserte land har man allerede mange års erfaring med å screene for flere titall forskjellige tilstander der medisinsk intervensjon vil være nyttig (14). Et nytt norsk nyfødtscreeningprogram som inkluderer 19 sykdommer er foreslått av det nasjonale rådet for kvalitet og prioritering i helsetjenesten. Problematiske sider ved en slik utvidelse av nyfødtscreeningen, blant annet hensynet til frivillighet og reelt samtykke, har vært drøftet i Tidsskriftet (15).
Mange involverte – manglende koordinering
Slik sykehistorien over illustrerer kan oppfølging av nevrodegenerativ sykdom hos barn være svært krevende. Den omfattende kontaktflaten – ikke bare med helsepersonell, men med alle typer hjelpere – stiller store krav til samarbeid. At over 10 – 15 fagpersoner i spesialisthelsetjenesten til enhver tid er involvert rundt én pasient er heller normen enn unntaket! I tillegg er det et like stort antall hjelpere i førstelinjetjenesten. Hvis man legger til at familiene til flere av disse pasientene ikke har røtter i Norge og ikke har norsk som morsmål, begynner bildet å bli enda mer realistisk.
Det er i praksis svært vanskelig å få til en helt tilfredsstillende koordinert oppfølging. Siden sykdommen kan endre seg raskt, kan det være vanskelig å forutsi den enkelte pasients behov. Barnehabiliteringen er ansvarlig for koordineringen av det medisinske tilbudet til pasienten og familien. Et sentralt poeng i oppfølgingen er å sikre at de involverte aktørene kontinuerlig blir oppdatert om situasjonen. Enkle rutiner, som det at man alltid må sende kopi av polikliniske notater til foresatte og fastlege, svikter lett. Individuell pasientplan med pasientkoordinator i førstelinjetjenesten er svært nyttig. Fastlegen er en uvurderlig samarbeidspartner. Stadig dokumentasjon når det gjelder sosiale rettigheter, omsorgslønn, pleiepenger og hjelpemidler er belastende for en familie som er hardt presset fra før. Frambu helsesenter er et landsdekkende kompetansesenter for sjeldne funksjonshemninger og har en viktig rolle i å støtte pasienten og dens familie.
Ny, kostbar behandling
Bortsett fra beinmargstransplantasjon, som har vært et behandlingstilbud i flere tiår ved utvalgte lysosomale avleiringssykdommer som Hurlers syndrom (mukopolysakkaridose type 1) (16), har behandlingsmulighetene vært få. Enzymerstatningsterapi (enzyme replacement therapy, ERT) har vært brukt siden tidlig i 1990-årene for enkelte lysosomale sykdommer, slik som Gauchers sykdom (17) og Fabrys sykdom (18), og behandlingsalternativer for andre tilstander har kommet til nesten hvert år. Også substratreduksjonsterapi (SRT) er et alternativ ved lysosomal sykdom (19), og i noen tilfeller kan substratreduksjonsterapi og enzymerstatningsterapi kombineres (20).
Det har vist seg at beinmargstransplantasjon kan stabilisere den demyeliniserende prosessen ved X-bundet adrenoleukodystrofi (X-ALD) hvis man starter behandlingen tidlig, helst før debut av nevrologiske symptomer (21). Nylig ble det rapportert gode resultater etter en mer raffinert form for genterapi hos to slike pasienter med normal intelligenskvotient og motorikk, men med demyeliniserende forandringer i hjernen (22). Ekstraherte hematopoetiske stamceller fra de to ble infisert med en virusvektor som var innsatt med villtypegenet for X-ALD. De transuderte (infiserte) stamcellene som ble injisert tilbake, påvirket gliacellene i hjernen slik at demyeliniseringen stanset opp. Motorikk og intelligenskvotient var fortsatt på normalt nivå etter to og tre års observasjonstid (22).
Bedrede behandlingsmuligheter er et sterkt argument for å innføre utvidet nyfødtscreening i Norge. Felles for de fleste nye behandlingsstrategier er at de isolert sett er kostbare. Enzymerstatningsterapi og substratreduksjonsterapi er livslang behandling. Det reiser spørsmålet om vi har råd til denne type behandling. For eksempel koster substratreduksjonsterapi med Miglustat ved Niemann-Picks sykdom type C hos en voksen pasient 2 millioner kroner i året (Torbjørn Ritzler, Actelion, Sverige, personlig meddelelse). Prisen er avhengig av pasientens vekt og blir derfor lavere for barn. Ved en kostnad-nytte-analyse må man imidlertid ta med i regnestykket potensielle besparelser gjennom reduserte offentlige utgifter til støttebehandling hvis pasienten responderer gunstig på behandlingen.
