En ung mann med multiple basalcellekarsinomer ble henvist til universitetssykehus på grunn av mistanke om et sjeldent syndrom. Å stille en korrekt sjelden diagnose kan være vanskelig, noe vi ønsker å belyse med denne kasuistikken.
En mann i slutten av 20-årene ble henvist til universitetssykehus fordi man mistenkte et sjeldent syndrom. Han hadde multiple basalcellekarsinomer, flere lokalisert til hodebunnen (fig 1).
Ifølge pasientjournalen var han født til termin etter et normalt svangerskap. Foreldrene var etnisk norske, friske og ubeslektede. Det ble bemerket på fødeavdelingen at gutten var stor – fødselsvekten var ca. 5 000 g, lengden 58 cm og hodeomkretsen 41 cm. Det store hodet ga mistanke om hydrocephalus. Gutten hadde ingen trykksymptomer. Han ble sendt til et universitetssykehus, og etter flere undersøkelser ble det planlagt operasjon. Dette ble stoppet da man mente at det allikevel ikke dreide seg om hydrocephalus, men cerebral gigantisme (Sotos’ syndrom).
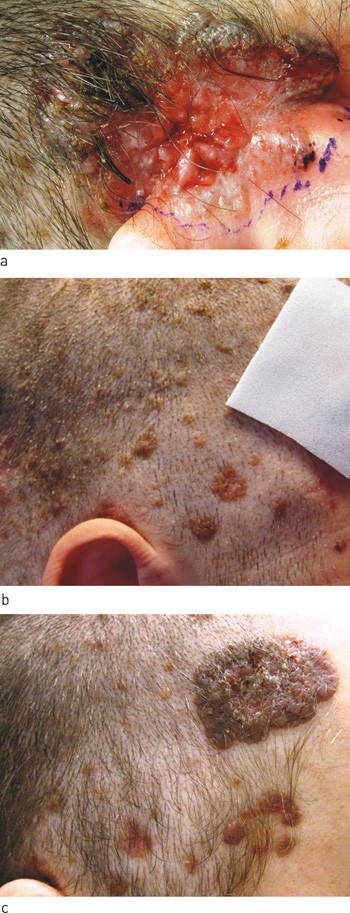
Figur 1 Pasienten ved første besøk hos hudlege. a) Væskende sår i basalcellekarsiom bak øret. b) Oversiktsbilde over basalcellekarsinomer i hodebunnen. c) Stort basalcellekarsinom i hodebunnen med sentral ulcerasjon og pigmentert rand
I journalen beskrives det ikke hvorfor man kom til denne konklusjonen, men historien viser at stort hode ikke alltid er hydrocephalus. Det kan være familiært eller ledd i et sjeldent syndrom, for eksempel Sotos’ syndrom, Gorlins syndrom, Cowdens syndrom, fragil X-syndrom, nevrofibromatose type 1, metabolske sykdommer eller andre tilstander.
Sotos’ syndrom er karakterisert av kraftig høydevekst og/eller stort hode, lærevansker av varierende grad og karakteristiske ansiktstrekk, som langt, smalt ansikt. Hjertefeil, nyrefeil og andre medfødte misdannelser kan være til stede. Mutasjoner i NSD1-genet er årsak til Sotos’ syndrom (1).
Sjeldne diagnoser bør, så sant det lar seg gjøre, bekreftes genetisk for å kunne si noe om prognose, arvegang og gjentakelsesrisiko. Uriktig diagnose kan få store konsekvenser.
Gutten ble fulgt opp regelmessig ved den lokale barneavdelingen. I barneskolealder ble han henvist til barne- og ungdomspsykiatrisk avdeling for ulike tester grunnet forsinket utvikling – dårlig talespråkutvikling og mangelfull fin- og grovmotorisk utvikling. Pedagogisk-psykologisk tjeneste fulgte gutten. Han hadde assistent og ekstratimer på skolen.
Han fikk jevnlig oppfølging av øyelege pga. langsynthet og periodevis innoverskjeling. Han ble fulgt av fysioterapeut og fikk sålebehandling for plattfot.
Da han var i tiårsalderen, oppdaget man at han hadde problemer med fingergrep på grunn av redusert fleksjon i ytterleddet på begge tomler. Manglende fleksjon ble påvist også i ytterleddet på begge stortær. Den ene tommelen ble operert, uten hell, og man valgte å avstå fra operasjon av den andre tommelen fordi det var «nokså atypiske forandringer».
På slutten av barneskolen var høyden 5 cm over 97,5-prosentilen, vekten var på 90-prosentilen, hodeomkretsen 4,5 cm over 97,5-prosentilen og skostørrelsen var 45.
Da han var i slutten av tenårene, fikk han hevelse og smerte i kjeven. Han ble operert for kjevecyster bilateralt i mandibula. Histologisk undersøkelse viste keratocyster (keratocystisk odontogen tumor).
Mannen oppsøkte fastlegen sin da han var i slutten av 20-årene fordi han ønsket å fjerne en brysom føflekk ved det ene øret. Fastlegen kjente lite til ham, ettersom han sjelden hadde tatt kontakt med legen. Han la merke til at pasienten hadde flere hudforandringer og stort hode og henviste ham til hudlege ved lokalsykehus.
Hudlegen fant flere basalcellekarsinomer hos pasienten (fig 1) og henviste til øre-nese-hals-lege og til øyelege for fjerning av lesjoner ved øre og øyelokk. Hudlegen og øre-nese-hals-legen fikk mistanke om Gorlins syndrom og henviste pasienten til universitetssykehus, der denne diagnosen ble stilt.
Pasienten hadde multiple basalcellekarsinomer som var vanskelig å behandle ved diagnosetidspunktet. Det ble påvist mutasjon i PTCH1-genet. Han ble vurdert av kjevekirurg og fikk påvist to kjevecyster som måtte opereres.
Diskusjon
Gorlins syndrom, også kalt nevoid basalcellekarsinomsyndrom, ble først beskrevet i 1960. De kliniske funnene var basalcellekarsinom, kjevecyster og bifide ribbein (2). I 1968 ble en norsk familie beskrevet (3). Senter for sjeldne diagnoser er nasjonalt kompetansesenter for diagnosen. De har registrert ca. 40 personer med Gorlins syndrom. Tilstanden er trolig underdiagnostisert i Norge. Forekomsten antas å ligge på rundt 1 : 30 000 (4), noe som skulle tilsi om lag 170 tilfeller her i landet.
Diagnosen stilles klinisk dersom pasienten fyller to hovedkriterier og ett tilleggskriterium eller ett hovedkriterium og tre tilleggskriterier (ramme 1) (5, 6). Dette er én av flere klassifiseringer (7, 8). Pasientene kan ha en rekke ulike medfødte misdannelser (8). Man finner mutasjon i PTCH1-genet hos 85 % (6). Arvegangen er autosomalt dominant. Diagnosen har høy penetrans og variabel ekspressivitet (6, 9). Nymutasjonsraten er over 50 % (4). Forventet livslengde antas å være over 70 år (10).
RAMME 1
Kriterier for klinisk diagnostisering av Gorlins syndrom (6). Diagnosen stilles klinisk dersom pasienten fyller to hovedkriterier og ett tilleggskriterium, eller ett hovedkriterium og tre tilleggskriterier.
Hovedkriterier
Multiple basalcellekarsinomer (mer enn fem i løpet av livet) eller ett basalcellekarsinom før 30 års alder
Kjevecyster
Palmare eller plantare groper (pits)
Forkalkning av hjernehinnen
Førstegradsslektning med diagnosen
Tilleggskriterier
Vertebra-/ribbeinsmisdannelse
Stort hode (> 97-prosentilen) med fremtredende panne
Hjerte- eller ovariefibrom
Medulloblastom
Lymfomesenteriske cyster eller pleuracyster
Leppe-gane-spalte
Preaksial eller postaksial polydaktyli
Øyeanomali (katarakt, utviklingsfeil, pigmentforandringer i retina)
Fastlegen observerte at det var noe ekstra med pasienten, som da altså var i slutten av 20-årene. Han henviste til spesialist, som fikk mistanke om Gorlins syndrom og henviste pasienten videre til universitetssykehus, der diagnosen ble bekreftet. En gjennomgang av sykehistorien illustrerer utfordringene med å gjenkjenne og stille en sjelden diagnose. En diagnose anses som sjelden når det er færre enn 100 kjente tilfeller per million innbyggere (11). I Norge lever det ca. 30 000 personer med sjeldne diagnoser (12), så samlet sett utgjør mennesker med sjeldne diagnoser en betydelig gruppe.
Stort hode, skjelettmisdannelser, øyeforandringer, lærevansker og kjevecyster er neppe tilfeldige sammentreff. Gutten hadde fått en sjelden diagnose, Sotos’ syndrom. Oralkirurgen som opererte kjevecystene, var kjent med at pasienten hadde denne diagnosen og koblet derfor ikke kjevecystene til det som senere viste seg å være den riktige diagnosen i dette tilfellet. Dessverre møtte ikke pasienten opp til kontroll hos oralkirurg.
Pasienten hadde lærevansker. Etter det vi kjenner til, er det ikke publisert studier hvor lærevansker hos pasienter med Gorlins syndrom er beskrevet (6). Det at pasienten hadde en sjelden diagnose fra før og hadde lærevansker, kan ha gjort at Gorlins syndrom ikke ble vurdert tidligere. Ved sjeldne tilstander hvor man ikke kan bekrefte diagnosen ved mutasjonsanalyse eller kliniske kriterier, bør pasienten revurderes etter noen år. Feil diagnose kan ha store konsekvenser for pasienten når det gjelder oppfølging og prognose, og for familien når det gjelder arv og gjentakelsesrisiko.
Det finnes ikke nasjonale utrednings-, behandlings- eller oppfølgingsrutiner for pasienter med Gorlins syndrom, men det finnes internasjonale retningslinjer, som vi anbefaler å følge (6, 13). Fastlegen bør bistå med koordinering av oppfølgingen.
Behandlingen er symptomatisk. Det er økt risiko for medulloblastom i de første tre leveår (14). Barnenevrolog bør følge pasientens psykomotoriske utvikling og vurdere behov for MR-undersøkelse. Barn og nydiagnostiserte voksne bør undersøkes for mulig øyeaffeksjon. Hjerte og eggstokker bør undersøkes for fibromer og opereres eller følges opp etter behov. Hudlege bør følge pasienten, da basalcellekarsinom er et vanlig symptom. Standard behandling av basalcellekarsinom er kirurgi, frysebehandling, fotodynamisk terapi (PDT), immunmodulerende krem eller laser.
Strålebehandling bør ikke benyttes hos pasienter med Gorlins syndrom, såfremt det kan unngås, da det er forbundet med økt risiko for stråleinduserte svulster og nye basalcellekarsinomer. Diagnostisk røntgen- eller CT-undersøkelse er også forbundet med slik risiko (15) og må brukes med forsiktighet. Strålebehandling av medulloblastom hos barn med syndromet har ført til stråleinduserte hjernesvulster og utallige basalcellekarsinomer (16, 17). Soleksponering uten tilstrekkelig beskyttelse gir økt risiko for basalcellekarsinom. Lite soleksponering fører til lavt nivå av vitamin D (18).
Pasientene må følges jevnlig med tanke på kjevecyster. Slike cyster kan bli store, og tennene kan bli forskjøvet (6, 8). Vi anbefaler at undersøkelse for kjevecyster gjøres med digitalt ortopantomogram (OPG) (fig 2) hos en kjeve-/ansiktsradiolog eller tannlege som er godt kjent med innstilling og tolking av denne undersøkelsen. Tannhelsekompetansesenteret (TAKO) ved Lovisenberg Diakonale Sykehus kan kontaktes ved spørsmål vedrørende tenner og kjeve.
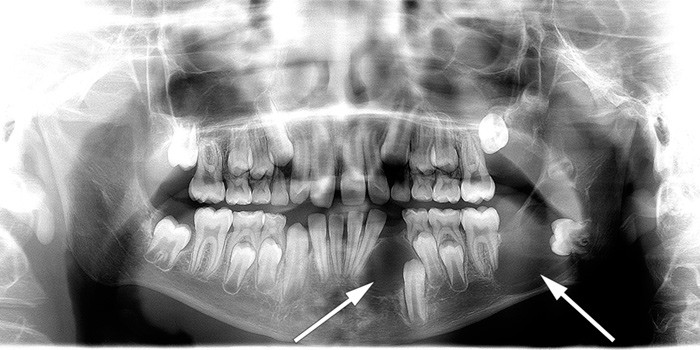
Figur 2 Illustrasjonsfoto av kjevecyster. Ortopantomografi (OPG) av gutt med diagnostisert Gorlins syndrom. På bildet ser man blandingstannsett med to store cystiske oppklaringer i mandibelen (piler). Histologisk undersøkelse viste keratocyster (keratocystisk odontogen tumor). Det er gitt samtykke til at bildet blir publisert
Behovet for psykososial oppfølging må vurderes (19). Pasient og familie bør få tilbud om henvisning til genetisk veiledning og informasjon om Senter for sjeldne diagnoser. I familier der genfeilen er kjent, anbefales prediktiv testing av førstegradsslektninger, også barn, fordi det har konsekvenser for videre oppfølging, forebygging og behandling (6, 13).
Det er nylig kommet et medikamentelt alternativ for behandling av basalcellekarsinomer når standard behandling ikke lenger er tilstrekkelig. Disse preparatene kalles «hedgehoghemmere» (ramme 2) (20). De virker ved å blokkere signalveien som, på grunn av genfeil i PTCH1, er aktivert ved utvikling av basalcellekarsinomer hos alle pasienter – ikke bare ved Gorlins syndrom (21, 22). Medikamentet er tilgjengelig som kapsel (vismodegib) og ble godkjent for klinisk bruk ved lokalavansert basalcellekarsinom i Norge i august 2013.
RAMME 2
«Hedgehoghemmere» hemmer signalveien «hedgehog» (pinnsvin) i cellene. Navnet «hedgehog» skriver seg fra forsøk med bananfluer, hvor fluene ble påført en spesifikk mutasjon. Mutasjonen medførte at fluelarvene ble små og hårete slik at de liknet pinnsvin. Senere har man vist at signalveien er sentral i utvikling av blant annet basalcellekarsinom. Hemming av signalveien kan benyttes terapeutisk ved basalcellekarsinom (20)
Oppsummering
Gorlins syndrom er en sjelden genetisk tilstand der pasientene kan utvikle medulloblastomer, kjevecyster og basalcellekarsinomer og ha medfødte skjelettforandringer. Udiagnostisert kan Gorlins syndrom innebære en rekke uheldige konsekvenser. Tidlig diagnose og god oppfølging er viktig for alle pasienter med sjeldne diagnoser.
Vi ønsker å gjøre leger og tannleger i Norge oppmerksomme på Gorlins syndrom slik at de, der det er mistanke om syndromet eller pasienten har diagnosen, henviser for utredning, behandling og oppfølging hos spesialister som kjenner diagnosen godt. Hudavdelinger ved universitetssykehusene og avdelinger for medisinsk genetikk er sentrale i utredning og oppfølging.
En nasjonal ressursgruppe for diagnosen er opprettet. Den består av hudlege, onkolog, genetiker, barnelege, spesialtannlege, øyelege, ortoped, plastikkirurg, kjevekirurg og rådgivere. Pasienter, pårørende og helsepersonell kan ta direkte kontakt med Senter for sjeldne diagnoser for informasjon om diagnosen og ved behov for kontakt med representanter i ressursgruppen.
